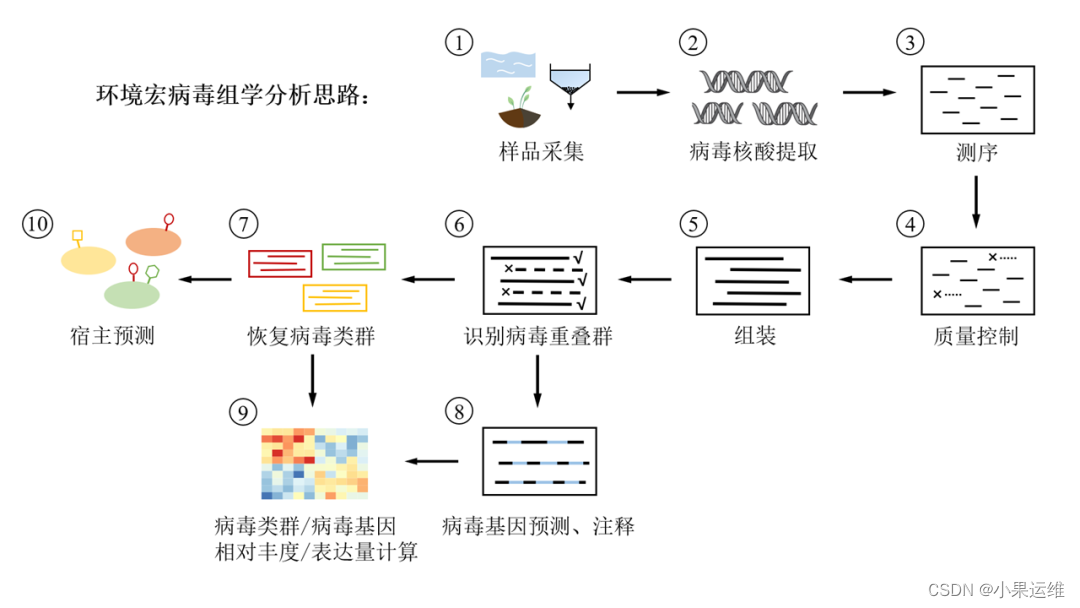
宏基因组学中基于Kraken2、Blast、ViPhOG和VirSorter等工具分析病毒组的详细流程,包括数据预处理、病毒序列检测、分类和功能注释等步骤

宏基因组学中分析病毒组的详细流程,包括数据预处理、病毒序列检测、分类和功能注释等步骤。
示例代码使用了常用的生物信息学工具,如Kraken2、Blast、ViPhOG和VirSorter等。
1. 数据预处理
质量控制:
使用FastQC进行原始测序数据的质量检查,然后使用Trimmomatic或Cutadapt去除低质量 reads 和接头。
步骤:
使用FastQC进行质量检查:
-
下载和安装FastQC:
- 访问FastQC的官方网站:Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data
- 下载并安装适合您操作系统的版本。
-
运行FastQC:
- 打开命令行界面(如终端或命令提示符)。
- 使用以下命令对您的FASTQ文件进行质量检查:
其中fastqc your_fastq_file.fastqyour_fastq_file.fastq是您的原始测序数据文件。
-
分析FastQC结果:
- FastQC将生成一个HTML报告,其中包含多个模块,如基本统计、序列质量分布、GC含量、接头内容等。
- 浏览报告并关注以下几个关键部分以确定数据质量:
- 序列质量:查看每个碱基位置的质量分布图,寻找质量下降的趋势。
- 接头内容:检查是否存在测序接头未被正确去除的情况。
- 碱基含量:检查GC含量是否在预期范围内。
使用Trimmomatic或Cutadapt去除低质量 reads 和接头:
这里我们将提供使用Trimmomatic和Cutadapt的示例。
使用Trimmomatic:
-
下载和安装Trimmomatic:
- 访问Trimmomatic的官方网站:USADELLAB.org - Trimmomatic: A flexible read trimming tool for Illumina NGS data
- 下载并解压Trimmomatic的jar文件。
-
运行Trimmomatic:
- 使用以下命令去除低质量 reads 和接头:
其中:java -jar \ path/to/trimmomatic.jar \ PE -phred33 \ your_fastq_file_R1.fastq \ your_fastq_file_R2.fastq \ trimmed_R1.fastq \ trimmed_R2.fastq \ unpaired_R1.fastq \ unpaired_R2.fastq \ ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 \ SLIDINGWINDOW:4:15 \ MINLEN:36your_fastq_file_R1.fastq和your_fastq_file_R2.fastq是你的双端测序数据文件。trimmed_R1.fastq和trimmed_R2.fastq是处理后的输出文件。unpaired_R1.fastq和unpaired_R2.fastq是无法配对的reads输出文件。ILLUMINACLIP:TruSeq3-PE.fa:2:30:10用于去除Illumina接头。SLIDINGWINDOW:4:15表示使用窗口大小为4,平均质量阈值为15的滑动窗口方法进行修剪。MINLEN:36表示保留至少36个碱基长度的reads。
- 使用以下命令去除低质量 reads 和接头:
使用Cutadapt:
-
下载和安装Cutadapt:
- 如果您使用的是Python环境,可以使用pip安装Cutadapt:
pip install cutadapt
- 如果您使用的是Python环境,可以使用pip安装Cutadapt:
-
运行Cutadapt:
- 使用以下命令去除低质量 reads 和接头(假设你有一个单端测序文件):
其中:cutadapt -a ADAPTER_SEQUENCE -q 15 -m 36 -o trimmed.fastq your_fastq_file.fastq-a ADAPTER_SEQUENCE指定接头序列。-q 15表示丢弃质量低于15的碱基。-m 36表示保留至少36个碱基长度的reads。trimmed.fastq是处理后的输出文件。your_fastq_file.fastq是你的原始测序数据文件。
- 使用以下命令去除低质量 reads 和接头(假设你有一个单端测序文件):
对于双端测序数据,你需要分别处理每一对R1和R2文件,并确保使用正确的接头序列。
组装
使用SPAdes、MEGAHIT或MIRA等工具进行短读长数据的组装。
使用SPAdes进行短读长数据的组装:
-
安装SPAdes:
- 如果您使用的是Linux或Mac OS,可以使用conda或pip进行安装:
conda install -c bioconda spades #或 pip install spades-python
- 如果您使用的是Linux或Mac OS,可以使用conda或pip进行安装:
-
运行SPAdes:
-
使用以下命令进行短读长数据的组装:
spades.py -1 reads_1.fastq -2 reads_2.fastq -o output_directory其中:
-1 reads_1.fastq和-2 reads_2.fastq是你的双端测序数据文件。-o output_directory指定输出目录。
-
如果你有单端数据,可以使用以下命令:
spades.py -s single_reads.fastq -o output_directory
-
-
分析结果:
- SPAdes会在输出目录下生成多个文件,包括组装后的 contigs(scaffolds)、contigs.fasta 文件以及一些统计信息。
使用MEGAHIT进行短读长数据的组装:
-
安装MEGAHIT:
- 如果您使用的是Linux或Mac OS,可以使用conda进行安装:
conda install -c bioconda megahit
- 如果您使用的是Linux或Mac OS,可以使用conda进行安装:
-
运行MEGAHIT:
-
对于双端数据,使用以下命令:
megahit -1 reads_1.fastq -2 reads_2.fastq -o output_directory其中:
-1 reads_1.fastq和-2 reads_2.fastq是你的双端测序数据文件。-o output_directory指定输出目录。
-
对于单端数据,使用以下命令:
megahit -r single_reads.fastq -o output_directory
-
-
分析结果:
- MEGAHIT会在输出目录下生成一个名为?
contigs.fa?的文件,其中包含了组装后的 contigs。
- MEGAHIT会在输出目录下生成一个名为?
使用MIRA进行短读长数据的组装:
-
安装MIRA:
- MIRA的安装通常需要下载并解压预编译的二进制文件,然后将其添加到系统路径中。具体步骤请参考MIRA的官方文档。
-
运行MIRA:
-
对于双端数据,创建一个名为
mira_config.txt的配置文件,内容如下:PROJECT = your_project_name SAMPLE = your_sample_name SETTING = denovo, small, normal, accurate TECHNOLOGY = illumina INPUT_TYPE = paired LIBRARY_TYPE = standard STRANDNESS = forward READ_GROUP_NAME = your_read_group_name FORWARD_READS = reads_1.fastq REVERSE_READS = reads_2.fastq OUTPUT_DIR = output_directory然后运行以下命令:
mira --config mira_config.txt -
对于单端数据,将配置文件中的
INPUT_TYPE改为single,并将REVERSE_READS行删除,然后运行相同的命令。
-
-
分析结果:
- MIRA会在输出目录下生成多个文件,包括组装后的 contigs(scaffolds)以及其他一些中间文件和统计信息。
2. 病毒序列检测
初步鉴定
使用Kraken2或者Centrifuge等工具进行宿主去除和病毒序列的初步鉴定。
使用Kraken2进行宿主去除和病毒序列鉴定:
-
数据准备:
- 确保你已经获得了原始的测序数据,通常是FASTQ格式。
- 如果数据是双端测序,需要将两个文件合并为一个(对于某些工具可能不需要)。
-
数据库准备:
- 下载或构建包含宿主和病毒参考序列的Kraken2数据库。这通常包括NCBI或其他来源的完整基因组序列。
- 使用
kraken2-build工具创建数据库,例如:kraken2-build --download-taxonomy kraken2-build --download-library viral --use-ftp kraken2-build --build --threads <num_threads> --db <database_name> viral/
-
运行Kraken2:
- 使用以下命令对测序数据进行分类和鉴定:
其中:kraken2 --db <database_name> --report <output_report> --threads <num_threads> --min-confidence 0.5 --classified-out <classified_output> <input_reads>--db <database_name>指定使用的数据库。--report <output_report>生成分类报告。--threads <num_threads>设置并行线程数。--min-confidence 0.5设置最小分类置信度阈值。--classified-out <classified_output>输出分类结果到指定文件。<input_reads>是你的输入测序数据文件。
- 使用以下命令对测序数据进行分类和鉴定:
-
宿主去除和病毒序列提取:
- 分析Kraken2的输出报告和分类结果,识别出被分类为宿主的reads并从原始数据中去除。
- 提取被分类为病毒的reads,这些就是初步鉴定的病毒序列。
使用Centrifuge进行宿主去除和病毒序列鉴定:
-
数据准备:
- 同样确保你已经获得了原始的测序数据。
-
数据库准备:
- 下载或构建包含宿主和病毒参考序列的Centrifuge数据库。可以使用
centrifuge-build命令构建数据库:
其中centrifuge-build --name <database_name> --ref <reference_directory> --seq <sequence_file><reference_directory>包含fasta格式的参考序列文件,<sequence_file>是一个包含序列名称和分类信息的tsv文件。
- 下载或构建包含宿主和病毒参考序列的Centrifuge数据库。可以使用
-
运行Centrifuge:
- 使用以下命令对测序数据进行分类和鉴定:
其中:centrifuge -x <database_name> -1 <input_reads_1> [-2 <input_reads_2>] -o <output_prefix> --report-file <output_report> --threads <num_threads>-x <database_name>指定使用的数据库。-1 <input_reads_1>和-2 <input_reads_2>分别指定单端或双端测序数据文件。-o <output_prefix>指定输出文件的前缀。--report-file <output_report>生成分类报告。--threads <num_threads>设置并行线程数。
- 使用以下命令对测序数据进行分类和鉴定:
-
宿主去除和病毒序列提取:
- 分析Centrifuge的输出报告,识别出被分类为宿主的reads并从原始数据中去除。
- 提取被分类为病毒的reads,这些就是初步鉴定的病毒序列。
病毒预测
使用VirSorter进行嵌段式和整合式病毒的预测。
首先,确保你已经安装了VirSorter和其依赖软件(如Python、BLAST+、Prodigal等)。以下是在Linux环境下使用VirSorter的基本步骤:
-
数据准备:
- 确保你已经获得了微生物基因组的fasta文件。
-
下载和安装VirSorter:
- 从GitHub上克隆VirSorter的代码库:
git clone https://github.com/jiarong/VirSorter.git cd VirSorter
- 从GitHub上克隆VirSorter的代码库:
-
配置环境:
- 确保你的环境中已经安装了BLAST+和Prodigal。如果没有,可以按照以下命令安装:
conda install -c bioconda blast prodigal
- 确保你的环境中已经安装了BLAST+和Prodigal。如果没有,可以按照以下命令安装:
-
运行VirSorter:
- 使用以下命令运行VirSorter:
其中:python virsorter.py -i <input_genome_file> -o <output_directory> --db <database_directory>-i <input_genome_file>指定输入的微生物基因组fasta文件。-o <output_directory>指定输出结果的目录。--db <database_directory>指定VirSorter数据库的目录。如果你还没有下载数据库,可以使用以下命令下载并解压:wget http://huttenhower.sph.harvard.edu/virsorter/db_virsorter.tar.gz tar -xzf db_virsorter.tar.gz
- 使用以下命令运行VirSorter:
-
分析结果:
- VirSorter会在输出目录下生成多个文件,包括分类结果、注释信息和可视化图谱。
- 分析
virsorter_output/candidate_viral_proteins.faa和virsorter_output/summary.txt文件,可以获取到预测的嵌段式和整合式病毒的信息。
以下是使用VirSorter的一个完整示例脚本:
# 下载并安装VirSorter
git clone https://github.com/jiarong/VirSorter.git
cd VirSorter
# 安装依赖软件
conda install -c bioconda blast prodigal
# 下载VirSorter数据库
wget http://huttenhower.sph.harvard.edu/virsorter/db_virsorter.tar.gz
tar -xzf db_virsorter.tar.gz
# 运行VirSorter
python virsorter.py -i input_genome.fasta -o output_directory --db db_virsorter
# 分析结果
less output_directory/virsorter_output/summary.txt在这个脚本中,你需要将input_genome.fasta替换为你的微生物基因组fasta文件的路径,将output_directory替换为你希望保存输出结果的目录。运行这个脚本后,你可以在output_directory/virsorter_output目录下找到预测的结果。
3. 病毒分类注释
使用BLAST对比NCBI病毒数据库进行分类注释(fasta文件较大的话推荐先prodigal然后使用蛋白序列使用diamond做序列比对diamond大基因序列快速比对工具使用详解-包含超算集群多节点计算使用方法_diamond比对-CSDN博客)。
makeblastdb -in virus_database.fasta -dbtype nucl
blastn -query assembled_contigs.fa -db virus_database.fasta -outfmt 6 -max_target_seqs 1 -num_threads $THREADS -evalue 1e-5 > blast_results.txt或者使用ViPhOG (Viral Phage Orthologous Groups) 进行更精细的分类。
ViPhOG (Viral Phage Orthologous Groups) 是一个专门用于病毒和噬菌体蛋白质分类的系统。以下是如何使用ViPhOG进行更精细的分类:
-
获取ViPhOG数据库和工具:
- 访问ViPhOG的GitHub页面(https://github.com/bebatut/viphog)并下载最新的ViPhOG数据库和相关的Python脚本。
-
安装依赖库:
- 确保你的系统已经安装了Python和以下依赖库:
- Biopython
- HMMER
- Diamond
- CD-HIT
如果尚未安装,你可以使用以下命令进行安装(假设你已经安装了conda环境管理器):
conda install -c bioconda biopython hmmer diamond cd-hit - 确保你的系统已经安装了Python和以下依赖库:
-
准备蛋白质序列文件:
- 将你需要分类的蛋白质序列整理成一个或多个人工 fasta 文件。
-
运行ViPhOG分类:
- 使用以下命令运行ViPhOG分类脚本:
python viphog.py -i your_protein_sequences.fasta -d ViPhOG_database_directory -o output_directory其中:
-i your_protein_sequences.fasta?是你的蛋白质序列文件。-d ViPhOG_database_directory?是ViPhOG数据库的目录。-o output_directory?是输出结果的目录。
-
分析结果:
- 分类完成后,ViPhOG会在指定的输出目录下生成多个文件,包括比对结果、分类信息等。
- 主要关注?
output_directory/viphog_assignments.txt?文件,其中包含了每个蛋白质序列的ViPhOG分类信息。
-
解读分类信息:
- ViPhOG分类信息是以数字编码的形式表示的,每个数字代表一个特定的蛋白质家族或功能类别。
- 你可以通过查阅ViPhOG数据库中的描述文件来解析这些数字编码,了解每个分类的具体含义。
4. 病毒功能注释
使用InterProScan进行蛋白质结构域和功能注释。
-
安装InterProScan:
- 如果你还没有安装InterProScan,可以从其官方网站(https://www.ebi.ac.uk/interpro/download.html)下载并按照提供的说明进行安装。
-
准备蛋白质序列文件:
- 将你需要进行注释的病毒蛋白质序列整理成一个或多个人工fasta文件。
-
运行InterProScan:
- 使用以下命令运行InterProScan:
interproscan.sh -i your_protein_sequences.fasta -f tsv -dp -goterms -pa -o interpro_results.tsv其中:
-i your_protein_sequences.fasta?是你的蛋白质序列文件。-f tsv?指定输出格式为TSV(制表符分隔值),便于后续的数据处理和分析。-dp?启用深度搜索模式,以提高注释的覆盖率。-goterms?启用Gene Ontology (GO) 术语注释。-pa?使用并行处理以加速注释过程(需要根据你的系统配置调整并行线程数)。-o interpro_results.tsv?指定输出结果的文件名。
-
分析结果:
- InterProScan运行完成后,会在指定的输出文件(在这个例子中是
interpro_results.tsv)中提供详细的蛋白质结构域和功能注释信息。 - 这个文件通常包含以下列:
protein_name: 蛋白质名称或标识符。sequence_length: 蛋白质序列的长度。signature_library_match: 结构域或功能注释所基于的数据库或签名库。signature_acc: 结构域或功能注释的唯一标识符。signature_desc: 结构域或功能注释的描述。start: 结构域在蛋白质序列中的起始位置。end: 结构域在蛋白质序列中的结束位置。score: 结构域匹配的得分(如果适用)。status: 结构域匹配的状态(例如,"T"表示确定的匹配,"C"表示条件性的匹配)。go_terms: 相关的GO术语(如果可用)。
- InterProScan运行完成后,会在指定的输出文件(在这个例子中是
-
解读和可视化结果:
- 使用文本编辑器或数据处理工具(如Excel、R、Python等)打开和分析
interpro_results.tsv文件。 - 根据需要筛选和汇总结果,例如按病毒种类、结构域类型或GO术语分类。
- 可以使用专门的可视化工具(如Cytoscape、GSEA或定制的脚本)将结果可视化,以帮助理解蛋白质的功能分布和网络。
- 使用文本编辑器或数据处理工具(如Excel、R、Python等)打开和分析
或者使用 eggNOG-mapper 对比 eggNOG 数据库进行功能注释。
-
安装 eggNOG-mapper:
- 如果你还没有安装 eggNOG-mapper,可以从 GitHub(https://github.com/eggnogdb/eggnog-mapper)?下载并按照提供的说明进行安装。
-
下载 eggNOG 数据库:
- 访问 eggNOG 网站(http://eggnogdb.embl.de/version/latest/download/)下载最新的?eggNOG 数据库。
- 解压下载的数据库文件,并记住解压后的目录路径。
-
准备蛋白质序列文件:
- 将你需要进行功能注释的病毒蛋白质序列整理成一个或多个人工fasta文件。
-
运行 eggNOG-mapper:
- 使用以下命令运行 eggNOG-mapper:
emapper.py -i your_protein_sequences.fasta --output outdir --cpu 4 --mode s --Diamond blastp --orthologs best --evalue 1e-5 --annotation cut_ga --evolutionary-distance 0.7 --search-tool diamond --database eggNOG_database_directory其中:
-i your_protein_sequences.fasta?是你的蛋白质序列文件。--output outdir?指定输出结果的目录。--cpu 4?设置使用的CPU核心数(根据你的系统配置调整)。--mode s?选择快速搜索模式(适用于大规模数据集)。--Diamond blastp?使用Diamond进行蛋白质比对。--orthologs best?只保留最佳的同源群注释。--evalue 1e-5?设置Diamond比对的E-value阈值。--annotation cut_ga?启用GO和EC编号的注释。--evolutionary-distance 0.7?设置进化距离阈值。--search-tool diamond?指定使用Diamond作为搜索工具。--database eggNOG_database_directory?指定eggNOG数据库的目录。
-
分析结果:
- eggNOG-mapper运行完成后,会在指定的输出目录(在这个例子中是
outdir)中提供详细的蛋白质功能注释信息。 - 这个目录通常包含以下文件:
emapper.annotations:包含每个蛋白质的详细注释信息,包括同源群、GO术语、EC编号等。emapper.best_hits.tsv:包含每个蛋白质的最佳同源群注释。emapper.results.all_contigs.tsv:包含所有比对结果的汇总信息。
- eggNOG-mapper运行完成后,会在指定的输出目录(在这个例子中是
-
解读和可视化结果:
- 使用文本编辑器或数据处理工具(如Excel、R、Python等)打开和分析
emapper.annotations文件。 - 根据需要筛选和汇总结果,例如按病毒种类、GO术语或EC编号分类。
- 可以使用专门的可视化工具(如Cytoscape、 REVIGO、定制的脚本)将结果可视化,以帮助理解蛋白质的功能分布和网络。
- 使用文本编辑器或数据处理工具(如Excel、R、Python等)打开和分析
5. 病毒丰度和多样性分析
使用Bracken对Kraken2的结果进行校正,得到更准确的病毒丰度。
bracken -k viral_classification.kraken2 -o bracken_output -t $THREADS -c contig_lengths.txt使用UCHIME或vFam进行嵌段式病毒的去冗余。
使用UCHIME进行去冗余:
UCHIME主要用于检测和去除PCR和测序过程中的假阳性序列(如chimeras)。虽然它最初设计用于16S rRNA基因的分析,但也可以应用于其他类型的序列,包括病毒序列。以下是一个基本的UCHIME命令行示例:
usearch -uchime_ref query_sequences.fasta -db reference_sequences.fasta -strand both -uchimeout uchime_output.uc这里:
query_sequences.fasta?是你的待去冗余的病毒序列文件。reference_sequences.fasta?是你的参考序列文件,可以包含已知的病毒序列或其他相关序列。-strand both?表示在正反两条链上都进行去冗余分析。-uchimeout uchime_output.uc?指定输出结果的文件名。
UCHIME将生成一个.uc格式的输出文件,其中包含了可能的chimeric序列的信息。你可以根据这些信息进一步过滤或去除潜在的冗余序列。
使用vFam进行去冗余:
vFam是一个专门针对病毒家族进行分类和去冗余的工具。它基于HMM(Hidden Markov Model)模型,可以识别和分类病毒序列。以下是一个基本的vFam命令行示例:
vfam classify --input query_sequences.fasta --output classified_sequences.fasta --hmms_dir vfam_hmms_directory这里:
query_sequences.fasta?是你的待去冗余的病毒序列文件。classified_sequences.fasta?是输出的分类和去冗余后的序列文件。vfam_hmms_directory?是包含vFam HMM模型的目录。
vFam将对输入的序列进行分类,并将结果写入到classified_sequences.fasta文件中。在这个过程中,vFam会自动去除冗余的序列。
使用Alpha diversity和Beta diversity指标(如Chao1、Shannon指数、UniFrac等)进行病毒群落多样性的分析。
Alpha多样性分析:
Alpha多样性主要关注单一样本内的物种丰富度和均匀度。以下是一些常用的Alpha多样性指数及其计算方法:
-
Chao1指数:
- Chao1指数是一种估计样品中未观测到的物种数目的方法,反映了物种丰富度。
- 在R语言中,可以使用
vegan包中的estimateR函数计算Chao1指数。
-
Shannon指数:
- Shannon指数考虑了物种丰富度和相对丰度,反映了物种多样性。
- 在R语言中,可以使用
vegan包中的diversity函数计算Shannon指数。
Chao1:对于基于病毒序列的数据,你可能需要先将数据转换为OTU(Operational Taxonomic Unit)表或者物种丰度矩阵,然后才能使用estimateR函数。以下是一个基本的示例:
library(vegan)
# 假设你已经有了一个OTU计数矩阵otu_table,其中行是样本,列是OTUs
# 并且你已经有了一个样本分类信息的数据框sample_data
# 计算Chao1指数
chao1 <- estimateR(otu_table, method = "chao1")
# 将结果与样本分类信息合并
alpha_diversity <- cbind(sample_data, Chao1 = chao1)?
在这个例子中,otu_table是一个矩阵,其中每个元素表示对应样本中特定OTU的数量。estimateR函数的method参数设置为"chao1",以指示计算Chao1指数。
然而,如果你的数据是原始的病毒序列数据,你可能需要先进行一些预处理步骤,例如:
- 序列聚类:将相似的序列聚类为OTUs。
- 计算OTU丰度:统计每个样本中每个OTU的数量。
这些步骤通常可以通过使用其他生物信息学工具或R包(如DECIPHER、cluster等)来完成。一旦你得到了OTU计数矩阵,就可以使用estimateR函数计算Chao1指数了。
Shannon指数?以下是一个基本的R代码示例:
library(vegan)
# 假设你已经有了一个OTU计数矩阵otu_table,其中行是样本,列是OTUs
# 并且你已经有了一个样本分类信息的数据框sample_data
# 计算Chao1指数
chao1 <- estimateR(otu_table, method = "chao1")
# 计算Shannon指数
shannon <- diversity(otu_table, index = "shannon")
# 将结果与样本分类信息合并
alpha_diversity <- cbind(sample_data, Chao1 = chao1, Shannon = shannon)Beta多样性分析:
Beta多样性主要关注不同样本间的物种组成差异。以下是一些常用的Beta多样性指标及其计算方法:
- UniFrac距离:
- UniFrac距离考虑了物种之间的进化关系,可以用来比较不同样本间的群落结构差异。
- 在R语言中,可以使用
picrust或phyloseq包中的函数计算UniFrac距离。
以下是一个基本的R代码示例(使用phyloseq包):
library(phyloseq)
# 假设你已经有了一个包含OTU表、样本信息和树状图的phyloseq对象ps
# 计算UniFrac距离
unifrac_dist <- phyloseq::distance(ps, "unifrac")
# 进行PCoA分析以可视化样本间的Beta多样性差异
pcoa_unifrac <- ordinate(ps, method = "PCoA", distance = unifrac_dist)
# 绘制PCoA图
plot(pcoa_unifrac, type = "n")
points(pcoa_unifrac, col = sample_data$Sample_Group, pch = 16)在进行病毒群落多样性的分析时,需要注意以下几点:
- 病毒的分类和系统发育信息可能不如微生物清晰,这可能影响到Alpha和Beta多样性指标的解释和应用。
- 病毒的复制和传播方式可能与微生物不同,因此在选择和解释多样性指标时需要考虑到这些特性。
- 在处理病毒序列数据时,可能需要进行额外的质量控制和预处理步骤,如去除宿主序列、过滤低质量序列等。
6. 病毒网络分析和热点区域识别
使用VIPER或VIPRA构建病毒互作网络。
使用VIPER构建病毒互作网络:
VIPER (Virus-Host Interaction Prediction Engine) 是一个基于机器学习的工具,用于预测病毒与宿主之间的蛋白质相互作用。
-
数据准备:
- 准备病毒蛋白和宿主蛋白的氨基酸序列文件。
-
安装和运行VIPER:
- VIPER提供了一个用户友好的Web界面(https://viperdb.scripps.edu/),可以直接在线进行分析。
- 在VIPER网站上,上传你的病毒蛋白和宿主蛋白的fasta文件。
- 设置参数,如预测方法、置信度阈值等。
- 点击提交按钮,VIPER将进行预测并生成一个互作网络。
-
分析结果:
- VIPER会返回一个互作网络,其中包括病毒蛋白和宿主蛋白之间的预测相互作用。
- 可以下载结果,并使用网络分析工具(如Cytoscape)进行可视化和进一步分析。
使用VIPRA构建病毒互作网络:
VIPRA (Virus-Host Protein-Protein Interaction Prediction and Ranking Algorithm) 是一个基于深度学习的工具,用于预测和排名病毒与宿主之间的蛋白质相互作用。
-
数据准备:
- 准备病毒蛋白和宿主蛋白的氨基酸序列文件。
-
安装和运行VIPRA:
- VIPRA是一个命令行工具,需要在本地安装和运行。
- 下载VIPRA源代码并按照提供的说明进行安装。
- 使用以下命令运行VIPRA:
vipra predict --virus_proteins virus_proteins.fasta --host_proteins host_proteins.fasta --output output_file.txt其中:
--virus_proteins?指定病毒蛋白的fasta文件。--host_proteins?指定宿主蛋白的fasta文件。--output?指定输出结果的文件名。
-
分析结果:
- VIPRA会生成一个文本文件,其中包含了病毒蛋白和宿主蛋白之间的预测相互作用及其得分。
- 可以将这些相互作用数据导入到网络分析工具(如Cytoscape)中,构建和可视化互作网络。
使用HotNet或其他方法识别病毒热点区域。
HotNet是一种用于识别生物网络中显著子网络(或称为热点区域)的方法,它通常应用于基因组学和蛋白质组学数据的分析。在病毒研究中,尽管HotNet主要用于识别基因或蛋白质的相互作用网络中的热点,但其基本原理也可以扩展到识别病毒序列或功能区域的热点。
以下是一个使用HotNet或其他方法识别病毒热点区域的基本步骤:
使用HotNet识别病毒热点区域:
除了HotNet,还有其他一些方法可以用于识别病毒热点区域,例如:
-
数据准备:
- 收集与病毒相关的数据,这可能包括病毒基因或蛋白质的表达数据、突变数据、互作数据等。
- 将这些数据转换为可以输入到HotNet的格式,通常是基因或蛋白质列表及其相关度量(如表达水平、突变频率、互作强度等)。
-
构建网络:
- 使用收集的数据构建一个病毒相关的网络。在这个网络中,节点代表病毒的基因或蛋白质,边代表它们之间的关系(如共表达、物理互作、功能相关性等)。
-
运行HotNet:
- 安装并加载HotNet软件包(如果尚未安装)。
- 使用以下命令运行HotNet(具体参数可能会根据你的数据和需求进行调整):
from hottopix import hotnet result = hotnet(graph_file='virus_network.txt', weight_file='node_weights.txt', alpha=0.05, beta=0.9) -
其中:
graph_file?是你的网络文件,包含节点和边的信息。weight_file?是你的节点权重文件,包含每个节点的相关度量。alpha?和?beta?是HotNet算法的参数,用于控制显著性检测和聚类过程。
-
分析结果:
- HotNet将返回一个或多个显著的子网络(热点区域),这些子网络中的基因或蛋白质可能在病毒的某个生物学过程中起着关键作用。
- 可以进一步分析这些热点区域中的基因或蛋白质的功能和相互作用,以了解其潜在的生物学意义。
-
Mutational Significance in Non-coding Regions (MiSNR):这种方法用于识别非编码区的突变热点,可以帮助揭示病毒调控元件的重要区域。
-
Gene Set Enrichment Analysis (GSEA):这种方法通过比较不同条件下的基因表达谱,可以识别在特定生物学过程中显著富集的基因集,这些基因集可能对应于病毒活动的热点区域。
-
Network Topology-based Methods:除了HotNet,还有其他基于网络拓扑的算法,如MAGMA、GiGA、jActiveModules等,可以用于识别网络中的显著子网络或模块。
以上是一个大致的宏基因组学中分析病毒组的流程,具体的分析步骤可能会根据研究目标和数据特性进行调整。在实际操作中,需要注意软件版本的选择和参数的优化,以及结果的可视化和解读。
?
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:veading@qq.com进行投诉反馈,一经查实,立即删除!