【Regulatory Genomics】Part3 GENOMICS AT NVIDIA、ATACWORKS
文章目录
来自Manolis Kellis教授(MIT计算生物学主任)的课
油管链接:Regulatory Genomics - Deep Learning in Life Sciences - Lecture 07 (Spring 2021)
本节课分为三个部分,本篇笔记是第三部分。
本节主要是介绍了英伟达在基因组学方面的一些工作,主要介绍了ATACWORKS这个模型。用于在有噪声的、有数据质量问题、分辨率低的数据,还原成清晰的测序数据。
Enchancing epigenomic data with deep learning

主要介绍了英伟达基因组学研究设计的一些领域,基于他们的硬件将机器学习、深度学习、加速计算应用在生物信息里
- 接下来主要是介绍这篇工作

- 首先介绍一下 ATAC-seq测序,

将reads贴上基因组上
用于测量染色质的可及性,通过DNA测序技术。
高测序读取覆盖率的“峰值”对应于基因组中开放的染色质区域。
有助于识别活跃的调控元件,构建调控网络,并研究非编码变异的影响。
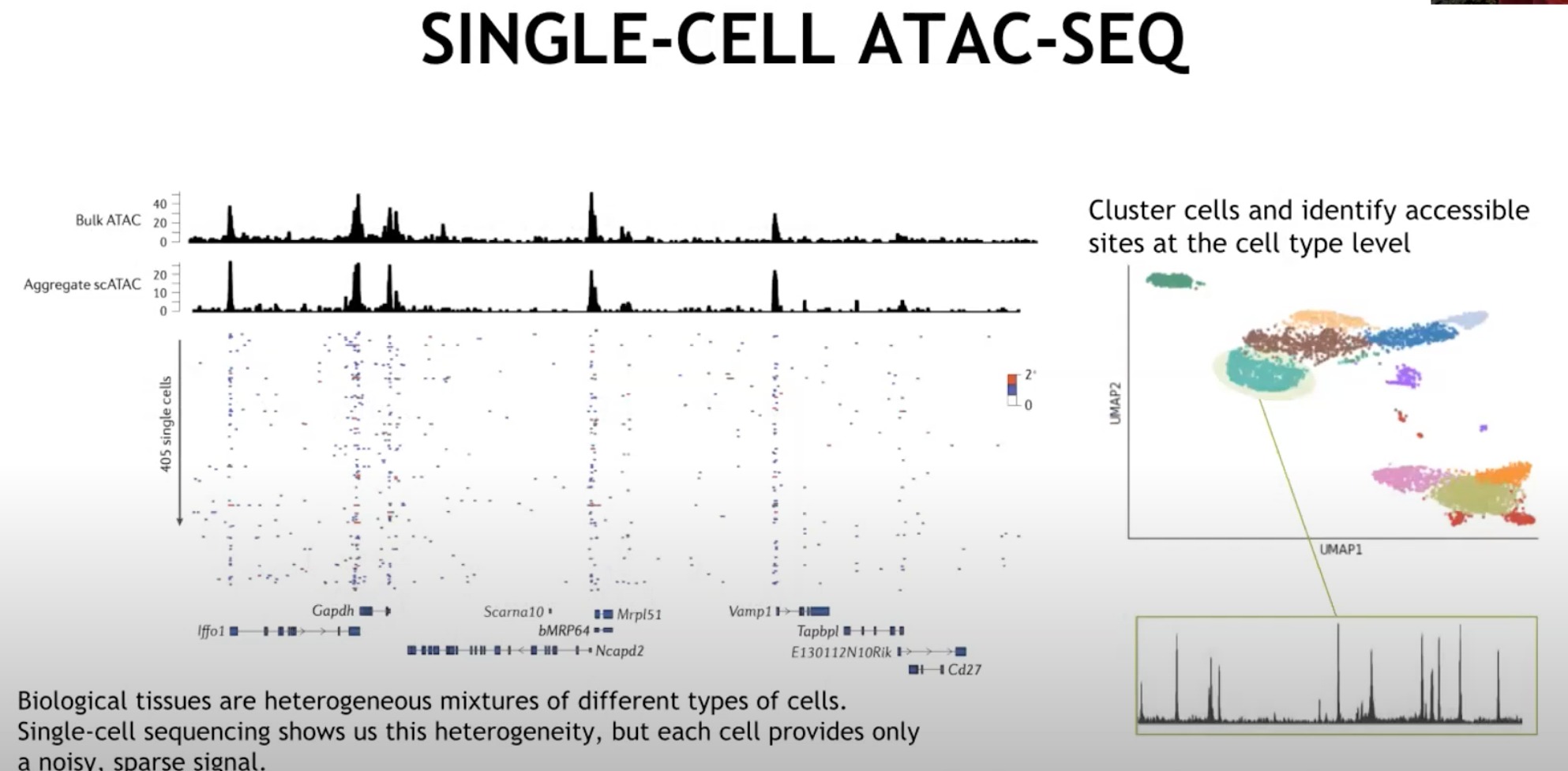
在单个细胞上进行测量,生物组织由不同类型的细胞混合组成,单细胞测序可以展示这种异质性,但每个细胞只提供有限的信号
-
Bulk ATAC:展示了大量细胞(不同种类细胞)的整体染色质可及性,平均数据。
-
Aggregate scATAC:这是多个单细胞ATAC-seq数据的聚合。尽管每个单独的细胞可能只提供有限的信息,但将它们聚合在一起可以提供与 Bulk ATAC 类似的信号。
- 下面的每行是一个细胞,如果不叠加在一起的话,是看不出什么东西的
-
所以我们使用如右侧所示的聚类,将细胞分类成一个个簇,需要哪个就提取一个簇出来,将其中的每个细胞信号叠加
UMAP - 是一种降维技术,用于将高维数据(如单细胞ATAC-seq数据)可视化到二维空间。
- 然后我们就可以比较为什么不同簇的细胞类型不同,为什么某些序列变异,影响的是这种细胞而非那种细胞
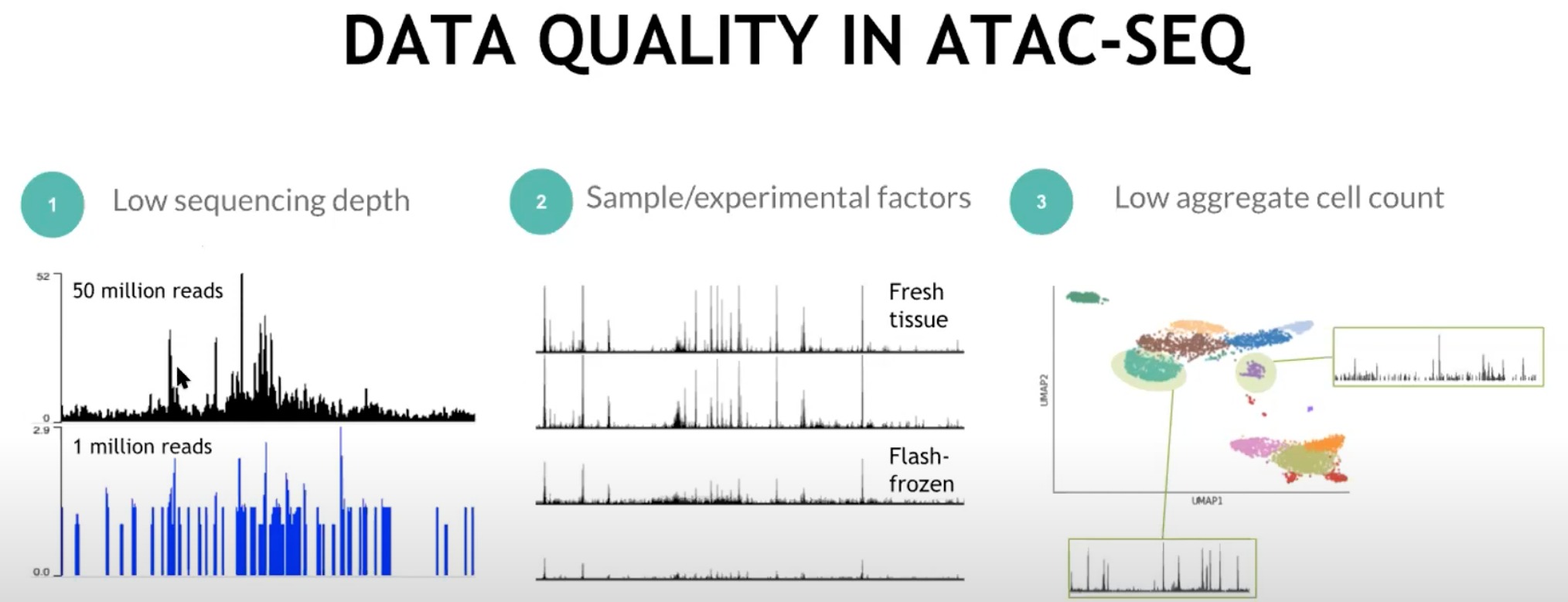
还是有些问题会导致ATAC-seq的数据质量问题
- Low sequencing depth(测序深度低):
- 这里展示了两种测序深度:50 million reads 和 1 million reads。
- 更高的测序深度可以提供更加精确且清晰的数据。
- Sample/experimental factors(样本/实验因素):
- “Fresh tissue”(新鲜组织)和“Flash-frozen”(快速冷冻)的样本。
- 不同的样本处理方式可能导致数据的差异。
- Low aggregate cell count(细胞计数低):
- 下面的柱状图展示了对应的ATAC-seq数据,突出显示了开放染色质区域的位置。
- 某一簇的细胞计数太低,那么结合起来后得到的数据可能不够代表性或精确。
AtacWorks: Improving the quality of ATAC-seq signals
1 model structure
基于以上的问题,提出了AtacWorks来增强ATAC-seq准确性,去除噪声

AtacWorks的功能:
- 它以 ATAC-seq 实验的coverage track为输入,并增强其准确性。
- 它还可以识别峰值,或称为开放染色质区域。
AtacWorks使用的特点(主架构是ResNet):
- 完全卷积模型
- 使用1-D卷积层:更适合于序列数据。对于基因组的每一个位置,有一个数值(读取数量)
- 扩张卷积: 这种卷积可以在保持参数数量不变的情况下增加感受野。
- 残差连接: 这有助于网络学习恒等函数,并防止深度网络中的梯度消失。ResNet特色
- 残差块(Residual Block)
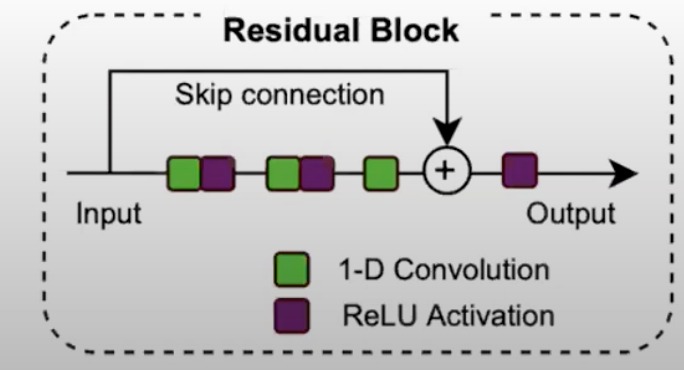
- Skip Connection(跳跃连接)
- 输入数据首先经过一个1-D卷积层,然后通过ReLU激活函数。
- 同时,输入数据也通过跳跃连接直接传递到输出端。
- 在块的末尾,卷积和激活的输出与原始输入相加(残差连接),然后产生输出。
- 残差块(Residual Block)
- 损失函数:回归和分类损失
- 回归:测量去噪覆盖轨迹的准确度
- 分类:对峰值位置进行分类的准确度。比如二分类,是否为峰值位置
- 这有助于模型同时学习连续的信号和峰值位置。
Why not!
有一个问题就是,为什么没有用基因组DNA的序列数据,而是使用coverage track数据。
是因为方便迁移,在一个细胞类型上训练的可以用在另一个细胞类型上。
因为如果输入dna序列的话,往往会学习到一些motifs跟染色体可及性相关的东西。而在一个细胞类型中的motif跟染色体可及性相关的性质,在另一种细胞类型中不一定也是这样。所以模型不一定能预测不同细胞类型的可及性。
当然这只是他们的选择而已,并不代表用dna序列就错
2 training strategy
AtacWorks利用深度学习方法从低质量或低覆盖度的ATAC-seq数据中预测出高质量的结果。这种方法能够在没有高覆盖度数据的情况下提取出有意义的生物信息。
在基因组测序中,覆盖率或深度是指某个基因组区域被测序读取覆盖的平均次数

- AtacWorks的学习方法是通过采取高覆盖度数据(例如全基因组测序数据),然后随机地对其进行下采样以获得噪声数据。
- 图中是从50million中选取1million
- 用这种方法,可以得到清晰的信号和噪声数据。之后,模型就可以在这两者之间进行训练,目的是从噪声数据中预测出清晰的结果。
-
Training and Inference
- 在Training部分(上半部分),噪声信号被用来训练AtacWorks模型,目的是使其能够从这种噪声信号中预测出清晰的结果。
- 在Inference部分(下半部分),模型被用来预测和增强其它来源的噪声数据。
- 例如,图中的“Sample B”展示了模型如何从噪声数据中恢复出去噪的信号和峰值预测。
-
输出 (Output)
- AtacWorks的输出是去噪后的信号和峰值调用。这可以应用于实际的生物信息学研究,使研究人员能够从低覆盖度的测序数据中获得有用的信息。
Performance of AtacWorks
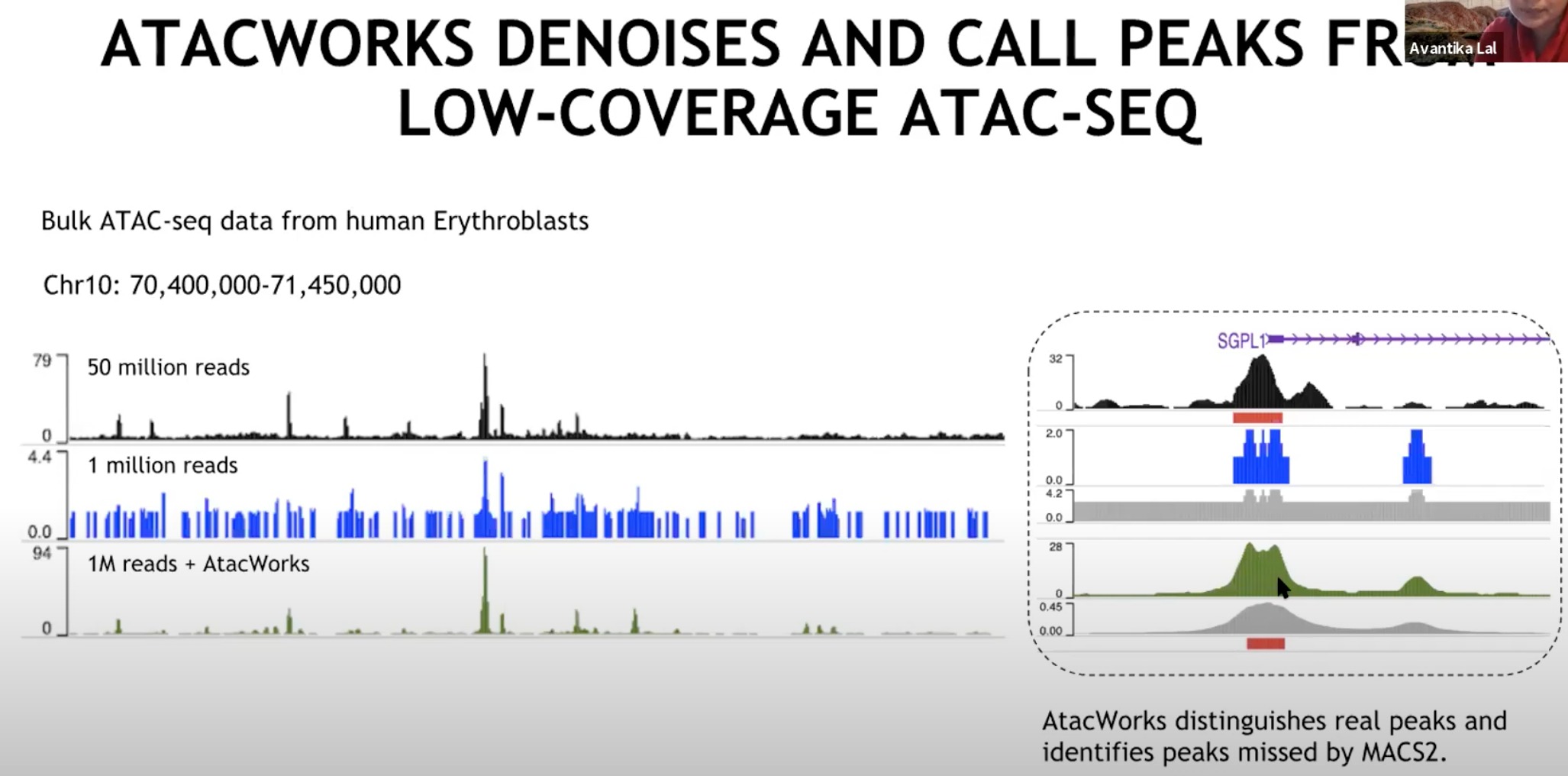
降低覆盖度后,数据会变得模糊,macs2无法识别。调用这个模型处理之后,去除背景噪声,就可以识别了。
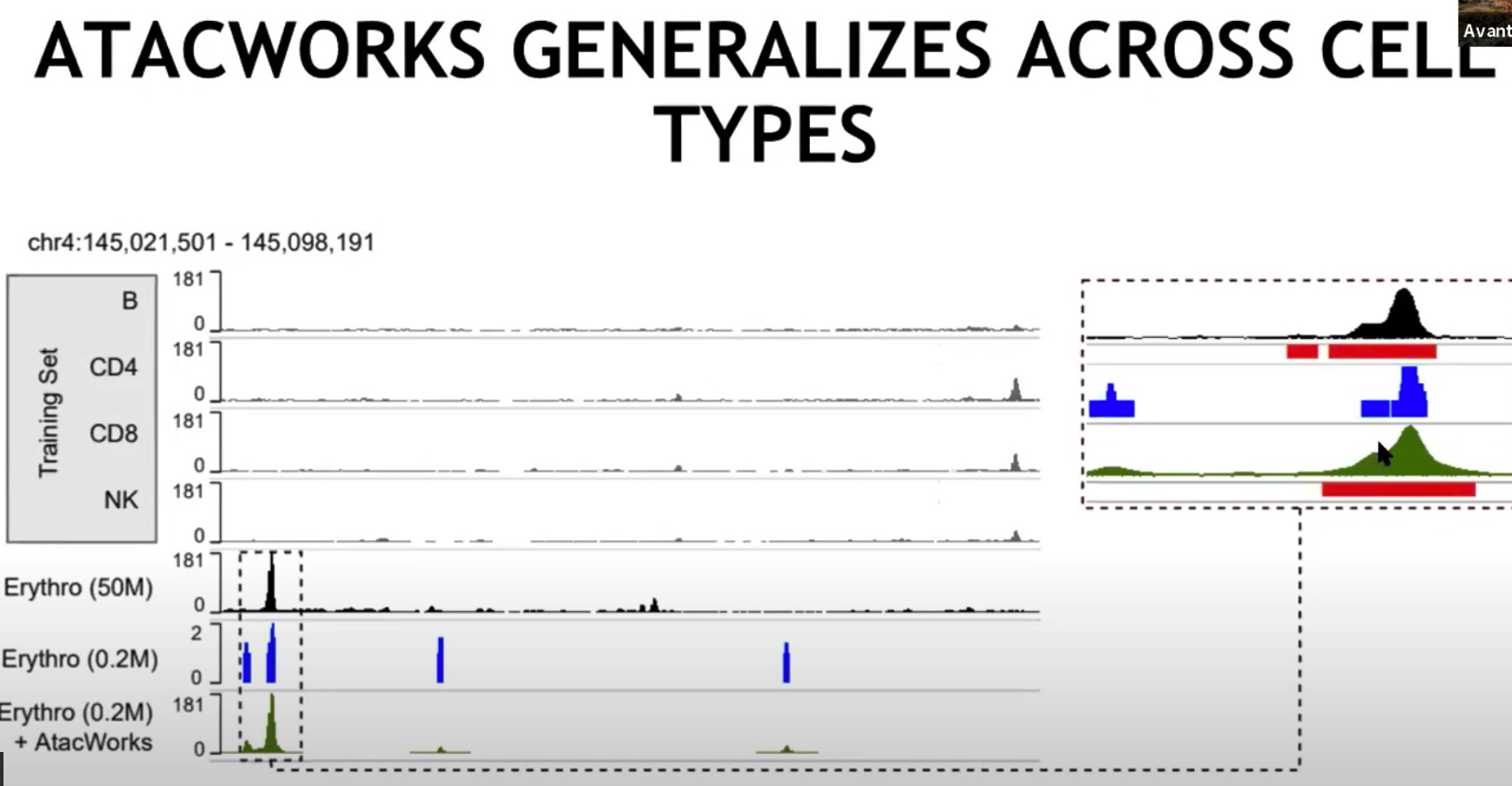
-
训练集
- 使用了更低的采样率
- B, CD4, CD8, NK:这些是训练集中的不同细胞类型。每个细胞类型的行显示了一个特定染色体区域(chr4:145,021,501 - 145,098,191)的ATAC-seq信号。这些信号图代表基因组某个区域的开放度或可及性。
-
对训练集中不存在的细胞类型也有作用

- 计算整个基因组范围内,子采样与原本数据的皮尔逊系数,发现经过模型处理过后的系数明显高
- Chr10,十号染色体是不存在在训练集中的。
- 效果也还不错
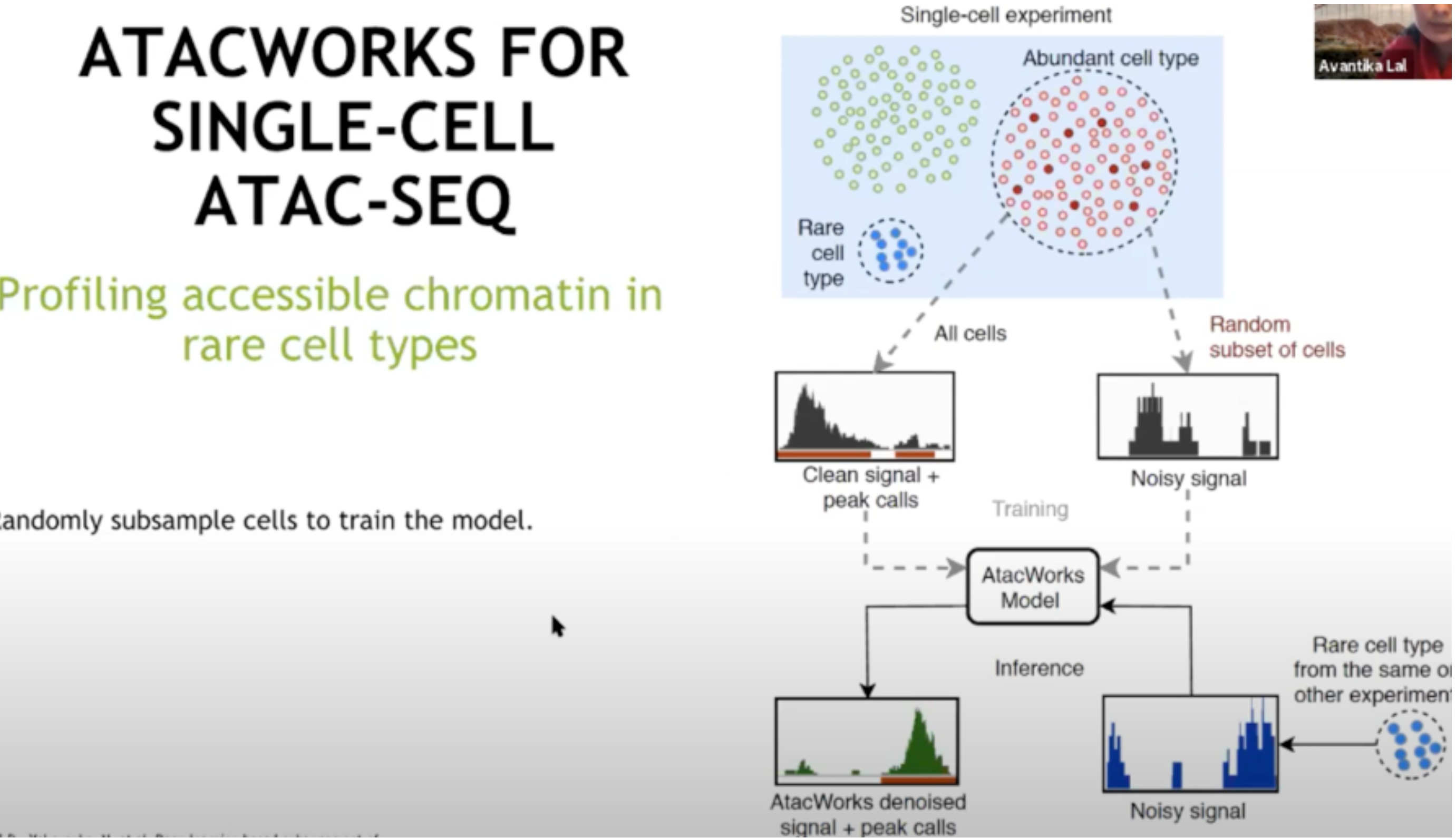
- 一个有趣的案例
- 用于细胞共济失调数据
- 如何在细胞类型混合中识别和分析那些数量较少的细胞
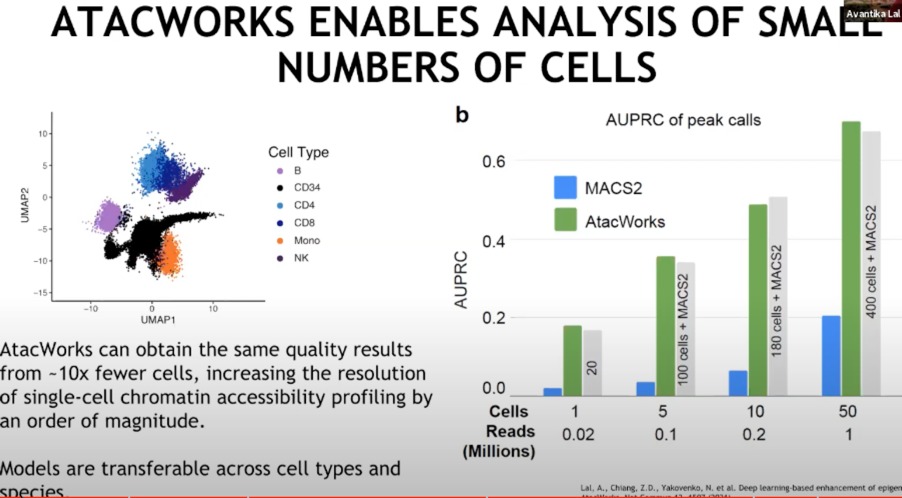
如何能够在处理少量细胞样本时提高分析的质量
- 图中的聚类表明,不同的细胞类型在基因组可及性方面具有不同的特征。
- 右侧 (b):显示了不同细胞样本量下,MACS2和ATAcWORKS在峰值调用性能上的比较。条形图显示了在不同数量的细胞和读取量(以百万为单位)下,ATAcWORKS比传统的MACS2工具在峰值调用上具有更高的准确性,特别是在样本数量较少时。
- ATAcWORKS可以使用比传统方法少10倍的细胞数量获得相同质量的结果,大幅提高了单细胞染色质可及性分析的分辨率。此外,这个模型可以跨不同的细胞类型和物种转移,这意味着一旦训练好,它可以应用于不同的生物学研究场景。
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:veading@qq.com进行投诉反馈,一经查实,立即删除!