身体的多组学特征识别出异质的健康表型和对生活方式干预的反应
今天给同学们分享一篇实验文章“Multiomic signatures of body mass index identify heterogeneous health phenotypes and responses to a lifestyle intervention”,这篇文章发表在Nat Med期刊上,影响因子为82.9。

结果解读:
Arivale队列特征
作者选择了一个由1,277名成年人组成的研究队列,他们参与了一个科学健康计划(Arivale),并且从同一次采血中进行了血浆代谢组学、蛋白质组学和临床实验室检测的耦合测量(图1a和方法)。这个研究设计使作者能够直接研究每个个体在BMI范围内的生理健康状态下组学平台之间的相似性和差异性。这个队列的特点是女性(64.3%),中年人(平均±标准差:46.6±10.8岁)和白人(69.7%)(扩展数据图1a-c和补充数据1)。根据世界卫生组织(WHO)对BMI截点的国际标准(体重过轻:<18.5 kg/m2,正常:18.5-25 kg/m2,超重:25-30 kg/m2,肥胖:≥30 kg/m2),基线BMI的患病率在正常、超重和肥胖类别之间相似,而只有0.8%的参与者属于体重过轻类别(体重过轻:10名参与者(0.8%),正常:426名参与者(33.4%),超重:391名参与者(30.6%),肥胖:450名参与者(35.2%))。在Arivale计划中,为所有参与者提供了个性化的健康生活方式辅导(方法),从而在多项健康指标上取得了临床改善。
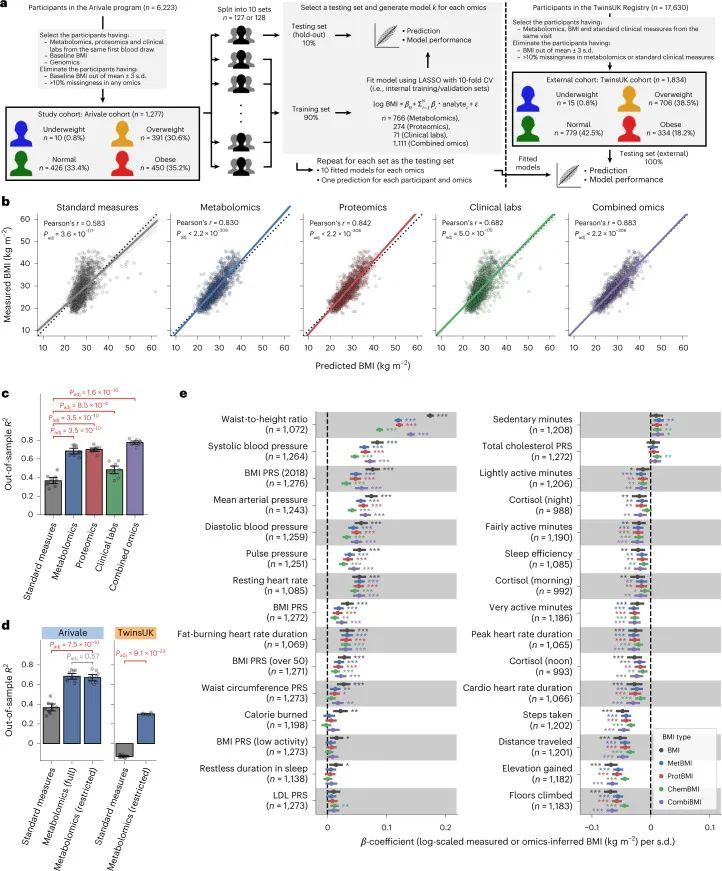
血液组学基于BMI模型
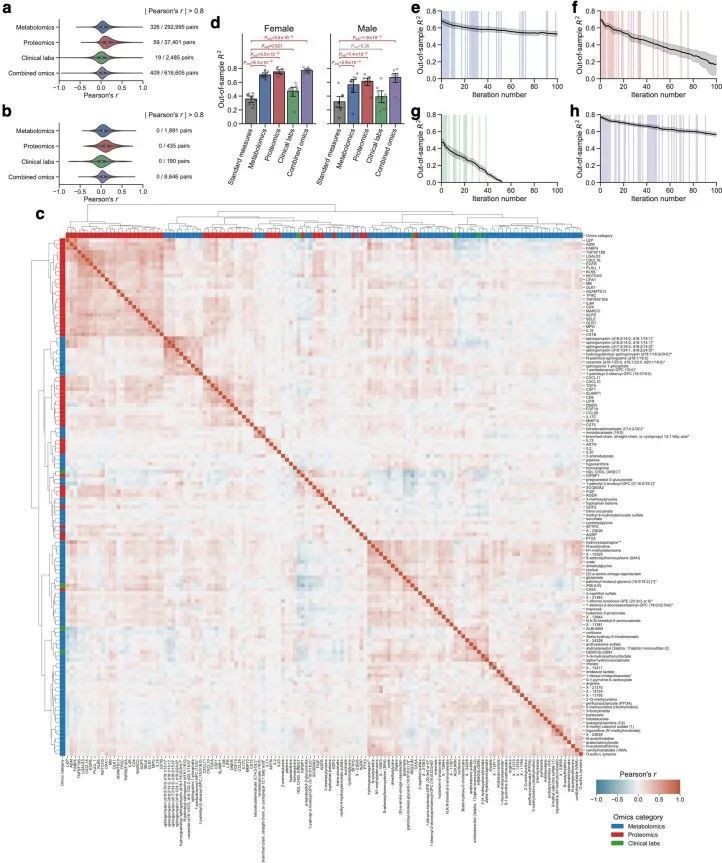
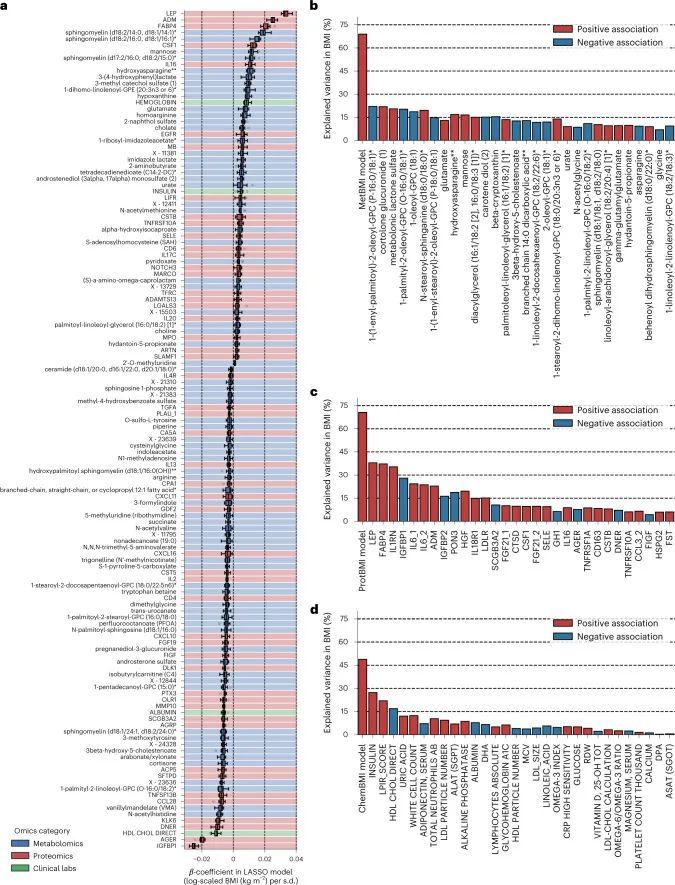
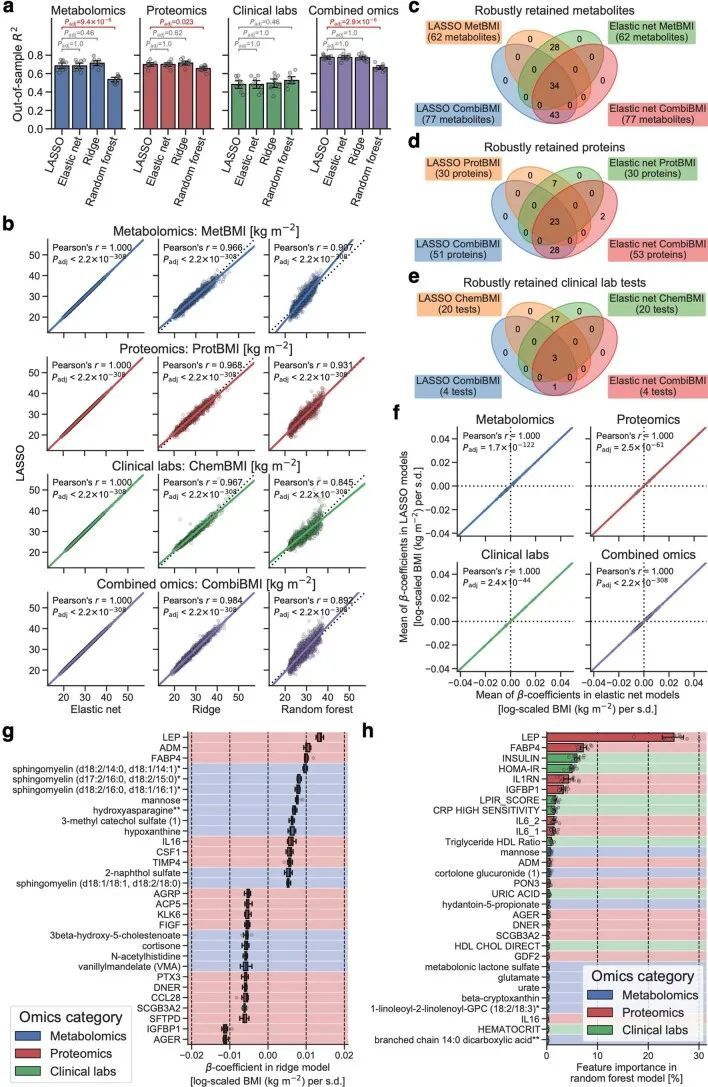
利用血浆分子分析的基线测量数据(766种代谢物、274种蛋白质和71种临床实验室检测项目;附加数据2),作者训练了机器学习模型来预测每个组学平台(代谢组学、蛋白质组学和临床实验室)或其组合的基线BMI:基于代谢组学的BMI(MetBMI)、基于蛋白质组学的BMI(ProtBMI)、基于临床实验室(化学检测)的BMI(ChemBMI)和基于组学的综合BMI(CombiBMI)模型。为了解决分析物之间的多重共线性问题(扩展数据图2a)并获得所有参与者的预测结果,作者采用了十折迭代的最小绝对收缩和选择算子(LASSO)算法进行十折交叉验证(图1a和方法)。这种方法为每个组学类别生成了十个拟合的稀疏模型(附加数据3),并为每个参与者从每个组学类别中生成了一个单一的测试(保留)集预测结果(图1b)。生成的模型在所有十个MetBMI、ProtBMI、ChemBMI和CombiBMI模型中分别保留了62个代谢物、30个蛋白质、20个临床实验室检测和132个分析物,这些模型之间的共线性较低(扩展数据图2b、c),符合LASSO算法的预期。与包括与肥胖相关的标准临床指标的模型(即包括性别、年龄、甘油三酯、高密度脂蛋白(HDL)胆固醇、低密度脂蛋白(LDL)胆固醇、葡萄糖、胰岛素和胰岛素抵抗的家庭模型评估(HOMA-IR)作为回归变量的普通最小二乘(OLS)线性回归模型;StandBMI模型)相比,每个基于组学的模型在BMI预测中表现出显著更高的性能,范围从外样R = 0.48(ChemBMI)到0.70(ProtBMI),而StandBMI模型为0.37(图1c)。CombiBMI模型在BMI预测中表现最佳(外样R = 0.78;图1c),1c),但所解释的差异并不完全是可加的,这表明尽管每个组学平台检测到的信号存在相当大的交集,但不同的组学测量仍然包含关于BMI的非冗余信息。此外,这些结果在性别分层模型中是一致的,除了男性ChemBMI模型的表现比StandBMI模型更好,但没有统计学意义(扩展数据图2d)。
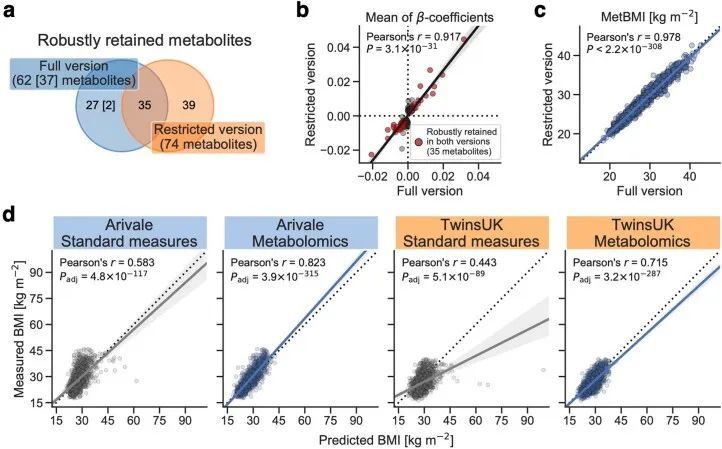
为了确认作者结果的普适性,作者调查了来自TwinsUK注册表的1,834名成年人的外部队列,其中包括血清代谢组学和前述的标准临床测量数据(图1a,扩展数据图1d-f和补充数据1)。作者使用适用于Arivale数据集的StandBMI和受限MetBMI模型计算了TwinsUK队列的BMI预测(扩展数据图3和方法)。与Arivale队列相比,受限MetBMI模型在TwinsUK队列上的绝对性能较低,但显著优于StandBMI模型(外样本R 2 = 0.30(MetBMI)和-0.13(StandBMI);图1d),确认血液代谢组学通常比标准临床测量更好地捕捉BMI。
基于组学的BMI模型中的预测特征
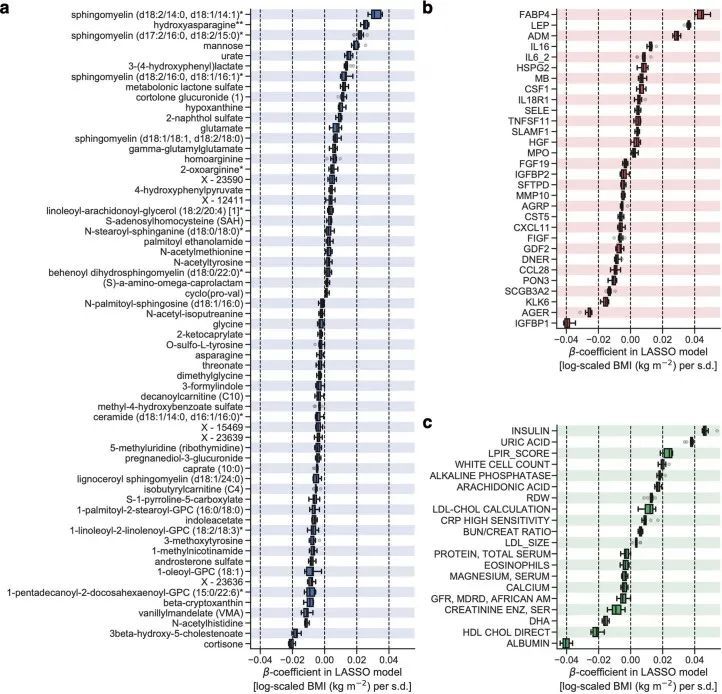
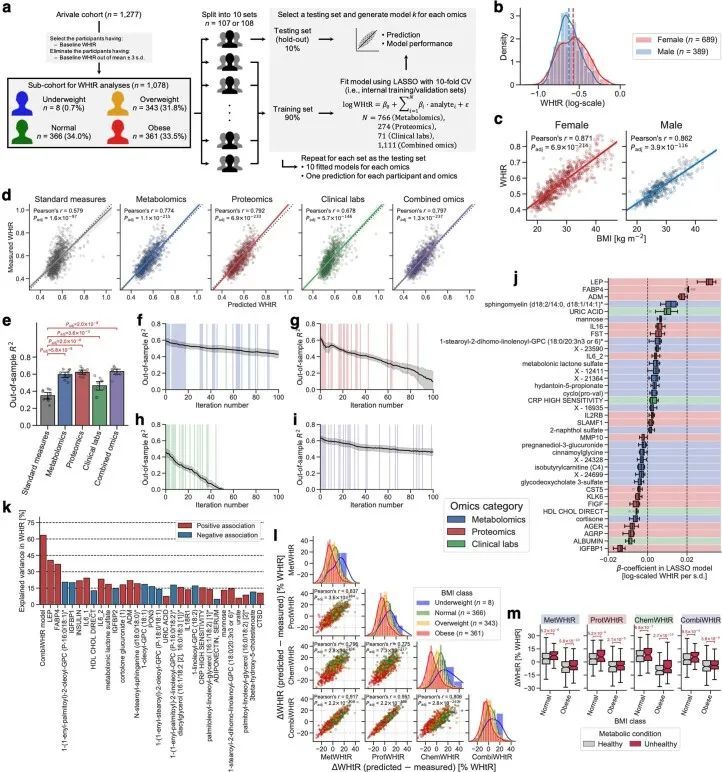
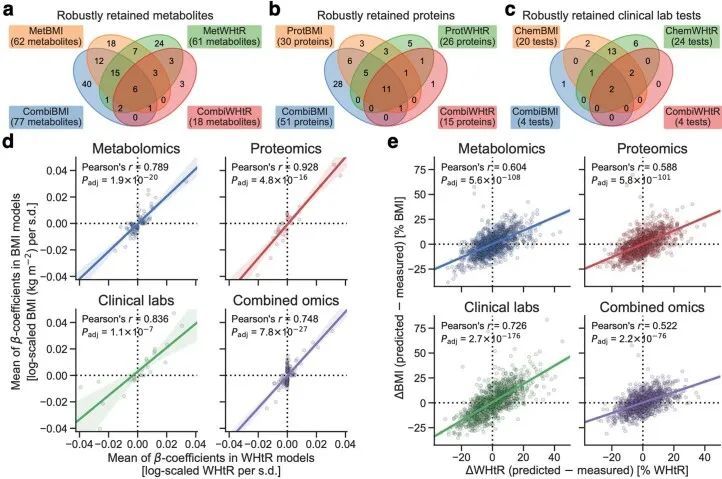
由于作者的LASSO线性回归模型在性能上与弹性网络、岭回归模型和非线性随机森林回归模型表现相似(扩展数据图4a、b),而且LASSO模型的β系数通常更容易解释,因此作者选择关注LASSO模型。然而,LASSO算法会随机保留高共线性组中的变量,并将其他变量的β系数设为0。为了确认变量选择过程的稳健性,作者在每次迭代结束时从输入组学数据集中删除最强的分析物(即具有十个β系数均值的绝对值最高的分析物),并进行LASSO建模的迭代。如果一个变量对模型是必不可少的,那么在删除它后,性能应该大幅下降。在所有组学类别中,首次5-9次迭代中观察到了样本外R 2 的急剧下降(扩展数据图2e-h),这表明至少在原始LASSO模型中具有最高绝对β系数的5-9个分析物对于预测BMI是必不可少的。与ProtBMI和ChemBMI模型相比,MetBMI模型中R 2 的整体斜率下降得更缓慢(扩展数据图2e-g),在所有十个LASSO模型中稳定保留的变量比例(扩展数据图图.5)5)与至少一个LASSO模型中保留的变量相比,MetBMI模型较低(MetBMI:62/209代谢物≈30%;ProtBMI:30/74蛋白质≈41%;ChemBMI:20/41临床实验室检测≈49%),这意味着代谢组学数据中关于BMI的冗余信息较其他组学数据更多。然而,代谢物仍占据了在所有十个CombiBMI模型中保留的132个分析物的58%(77个代谢物,51个蛋白质和四个临床实验室检测;图图.2a),这表明每个组学类别都具有关于BMI的独特信息。CombiBMI模型中最强的预测因子主要是蛋白质;具有平均绝对β系数>0的分析物。02年的正向预测因子是瘦素(LEP)、肾上腺髓质素(ADM)和脂肪酸结合蛋白4(FABP4),负向预测因子是胰岛素样生长因子结合蛋白1(IGFBP1)和高级糖基化终产物特异性受体(AGER;也称为RAGE)。这些最强的蛋白质在弹性网络模型中保持一致(扩展数据图4c-f),并且在岭回归和随机森林模型中具有很高的重要性(扩展数据图4g,h)。
标准BMI分类中的代谢异质性
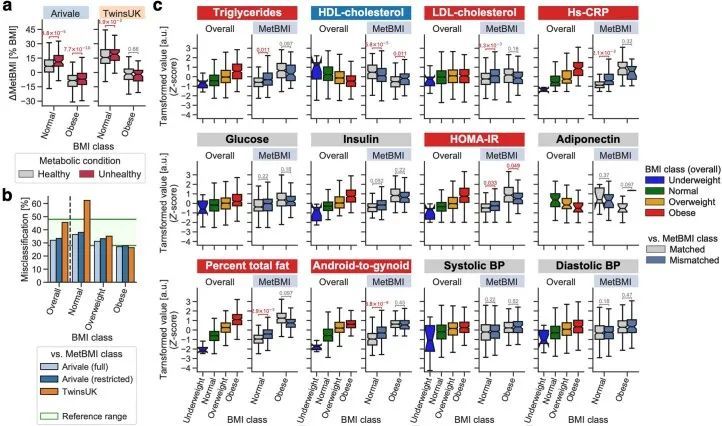
尽管基因组学推断的BMI与经典BMI显示出类似的表型关联(图1e),但作者观察到预测BMI与测量BMI之间的差异(ΔBMI)在基因组学类别之间高度相关,皮尔逊相关系数范围从0.64(ChemBMI与CombiBMI)到0.83(ProtBMI与CombiBMI)(图3a),这意味着这种偏差源于受扰动的生理状态的真实生物信号,而不是噪音或建模误差。当根据世界卫生组织的国际标准将正常和肥胖的BMI类别细分为代谢健康的临床定义(即,如果存在两个或更多代谢综合征风险则定义为代谢不健康;方法) 34,35 ,对于所有基因组学类别,MUNW和MUO组的ΔBMI显著高于代谢健康的正常体重(MHNW)和代谢健康的肥胖(MHO)组(图3b),这表明模型预测的偏差与代谢健康有关。

然而,对于代谢健康并没有普遍接受的定义。鉴于基因组学推断的BMI具有很高的可解释性和直观性,作者探索了一个潜在的应用:使用基因组学推断的BMI(而不是实际的BMI)来改进对肥胖和代谢健康的分类,符合世界卫生组织的国际标准。根据标准的BMI截断点,每个参与者根据测量和基因组学推断的BMI被分类,并且当测量的BMI类别与每个基因组学推断的BMI类别匹配或不匹配时,被分为匹配组或不匹配组。对于所有基因组学类别和BMI类别,与基因组学推断的BMI类别相比,误分类率约为30%(图3c),与先前报道的心脏代谢健康分类的误分类率一致。然后,作者研究了基因组学基础的误分类与正常或肥胖BMI类别之间的关系,以及与肥胖相关的临床血液标志物(补充数据6)之间的关系,包括甘油三酯、高密度脂蛋白胆固醇、低密度脂蛋白胆固醇、高敏C-反应蛋白(hs-CRP)、葡萄糖、胰岛素、HOMA-IR、糖化血红蛋白A1c(HbA1c)、脂联素和维生素D 3,15,23,38,39 。由于ChemBMI和CombiBMI模型与这些标志物不独立,因此在此分析中仅研究了相对于MetBMI或ProtBMI类别的误分类。正常BMI类别的不匹配组显示出与BMI正相关的标志物(+ BMI )的显著较高值,包括甘油三酯、hs-CRP、葡萄糖和HOMA-IR,以及与BMI负相关的标志物(- BMI )的显著较低值,包括高密度脂蛋白胆固醇和脂联素,与正常BMI类别的匹配组相比(FDR < 0.05; 图 图.3d)。. 这些模式表明,被错误分类为正常BMI类别的参与者具有与超重或肥胖个体相比较不健康的分子特征,对应于具有MUNW表型的个体。相反,被错误分类为肥胖BMI类别的不匹配组与匹配组相比,正负BMI相关标记的值分别显著较低和较高(FDR < 0.05; 图 图.3d),这表明被错误分类为肥胖BMI类别的参与者具有与超重或正常体重个体相比较健康的血液特征,对应于具有MHO表型的个体。
作者重新检查了27个与BMI相关的数值生理特征(图1e和补充数据6),并发现在WHtR(+ BMI )、心率(+ BMI )、血压(+ BMI )和日常体力活动(- BMI )测量方面,匹配组和不匹配组之间存在显著的表型差异一致模式(FDR < 0.05;图3e)。匹配组和不匹配组之间的BMI PRS(+ BMI )没有差异(图3e),这意味着生活方式或环境因素可能与测量和组学推断的BMI之间的不一致相关,而不是遗传风险。此外,作者使用TwinsUK队列验证了这些发现(扩展数据图6)。综上所述,这些结果表明,组学推断的BMI与经典BMI及标准BMI截断点所捕捉的异质代谢健康状态相关。
腹部肥胖和基于组学的BMI模型
身体脂肪分布是了解肥胖异质性的重要特征。特别是腹部肥胖,其特点是在腹部区域存在过多的内脏脂肪(而不是皮下脂肪),与代谢综合征等慢性疾病有关。因此,作者使用与基因组学BMI模型相同的方案,分析了腰围臀围比(WHtR),这是一种衡量腹部肥胖的人体测量指标。基于基因组学的WHtR模型显示出一致的结果和特征,与基因组学BMI模型相似。此外,在TwinsUK队列中,安卓区域的脂肪DXA测量值在正常BMI类别中,不匹配组与匹配组相比显著较高。总的来说,尽管传统的BMI需要脂肪分布的补充信息来诊断腹部肥胖,但基于基因组学的BMI模型可能捕捉到了肥胖的特征,包括腹部肥胖。
肠道菌群和基因组学推断的身体质量指数
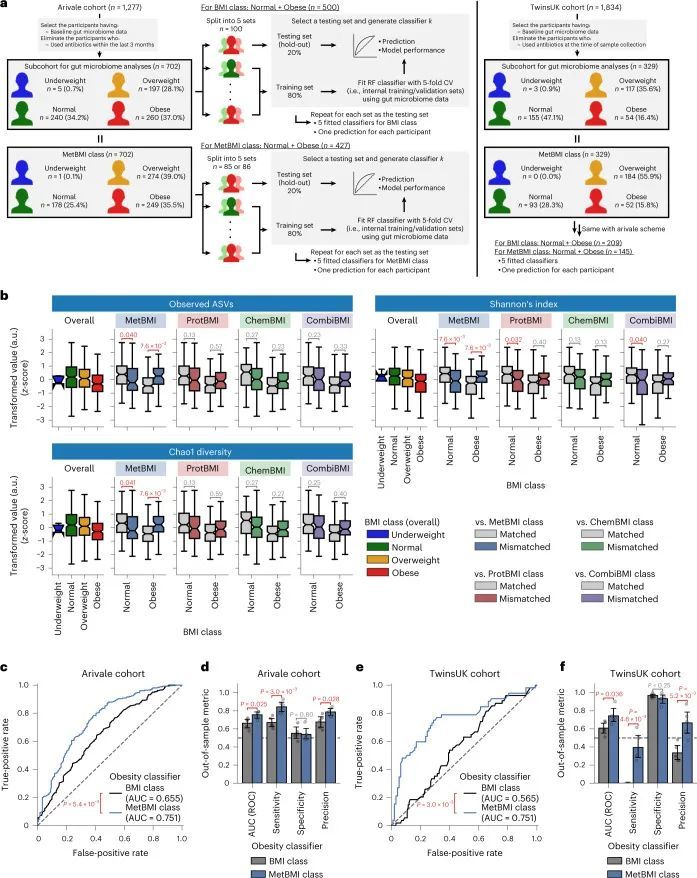
鉴于作者先前的发现,血液代谢物与细菌多样性之间的关联取决于BMI 20 ,以及当前的发现表明基于组学的BMI模型捕捉到了异质的代谢健康状态(图(图3)),作者假设MetBMI比实际BMI更好地代表了肠道菌群的α多样性。对于那些同时拥有粪便源肠道菌群和血液组学数据集的702名Arivale参与者(图(图4a4a和方法)),作者研究了肠道菌群α多样性(观察到的物种数量、Shannon指数和Chao1指数)与基于组学的BMI误分类之间的关系。与MetBMI类别匹配和不匹配的组之间,在正常和肥胖的BMI类别中,所有α多样性指标都显示出显著差异(图(图4b)),与与BMI呈负相关的表型模式一致(图3d,e),这意味着MetBMI类别更好地反映了细菌多样性,而不是标准的BMI类别。对于其他组学类别的误分类,在所有α多样性指标和BMI类别中都没有显示出这些显著差异(图(图4b),与作者之前的观察一致,即血浆代谢组学与肠道菌群结构的关联比蛋白质组学或临床实验室更强。20 。
基因组学推断的体质指数对生活方式干预的反应
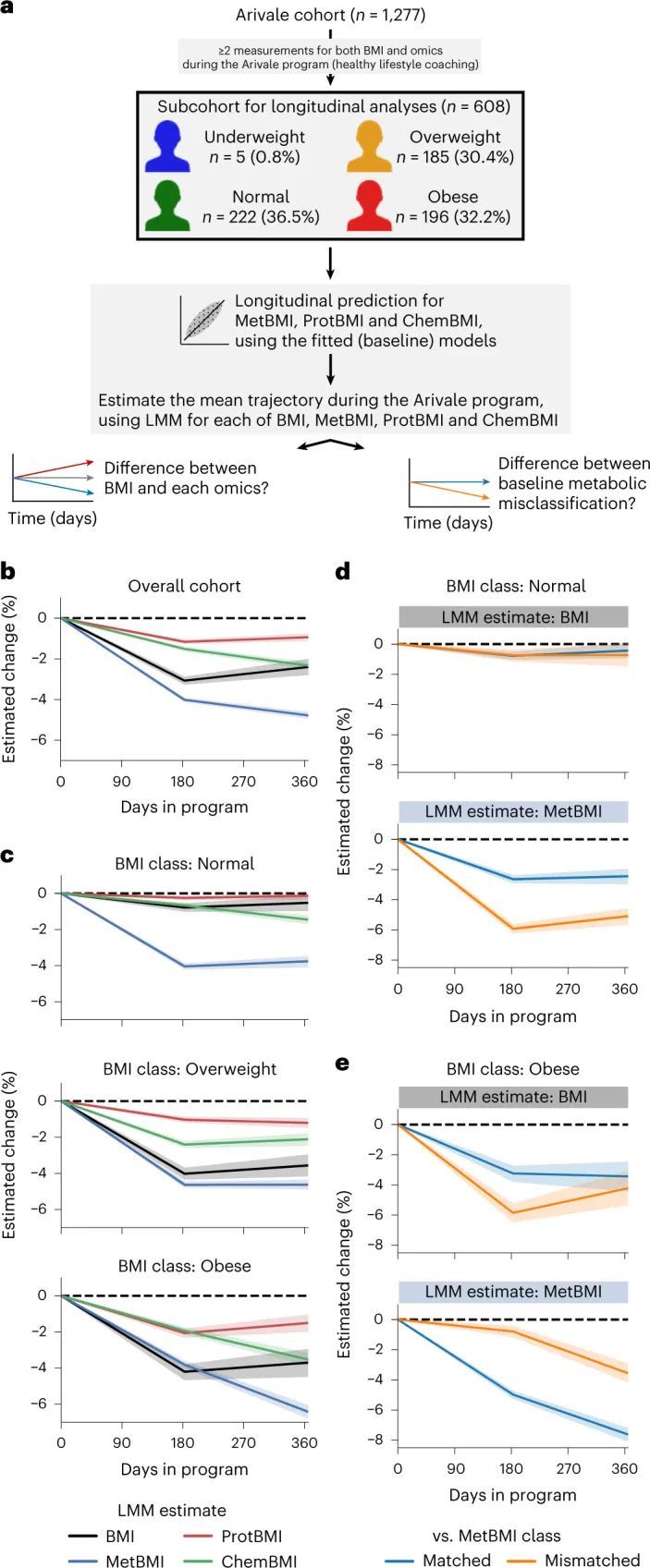
在Arivale计划期间,对608名参与者的组学数据进行了纵向变化的研究,基于可用的纵向测量数据(图(图5a)和方法)。鉴于数据收集的计数和时间点存在参与者相关的变异性,作者使用线性混合模型(LMM)估计了Arivale子队列中每个测量或组学推断的BMI的平均轨迹(方法中为每个参与者设置了随机效应)。与先前的分析一致,整体队列的平均BMI估计在计划期间减少了(图(图5b))。MetBMI的减少大于测量BMI的减少,而ProtBMI的减少最小,甚至小于测量BMI的减少(图(图5b)),这表明血浆代谢组学在短期内对生活方式干预非常敏感,而蛋白质组学(从同一次采血中测量)在同一干预期间更难改变。随后,作者根据基线BMI分类生成了LMM模型。测量的BMI、ProtBMI和ChemBMI的平均估计值在超重和肥胖的BMI类别中随时间呈负变化,但在正常的BMI类别中没有变化(图5c)。相反,平均的MetBMI估计值在所有BMI类别中显著下降(图5c),这表明代谢组学数据捕捉到了生活方式干预对代谢健康反应的信息,超越了基线BMI类别以及实际BMI和其他组学特征的变化。
血液分析物网络动力学和MetBMI分类
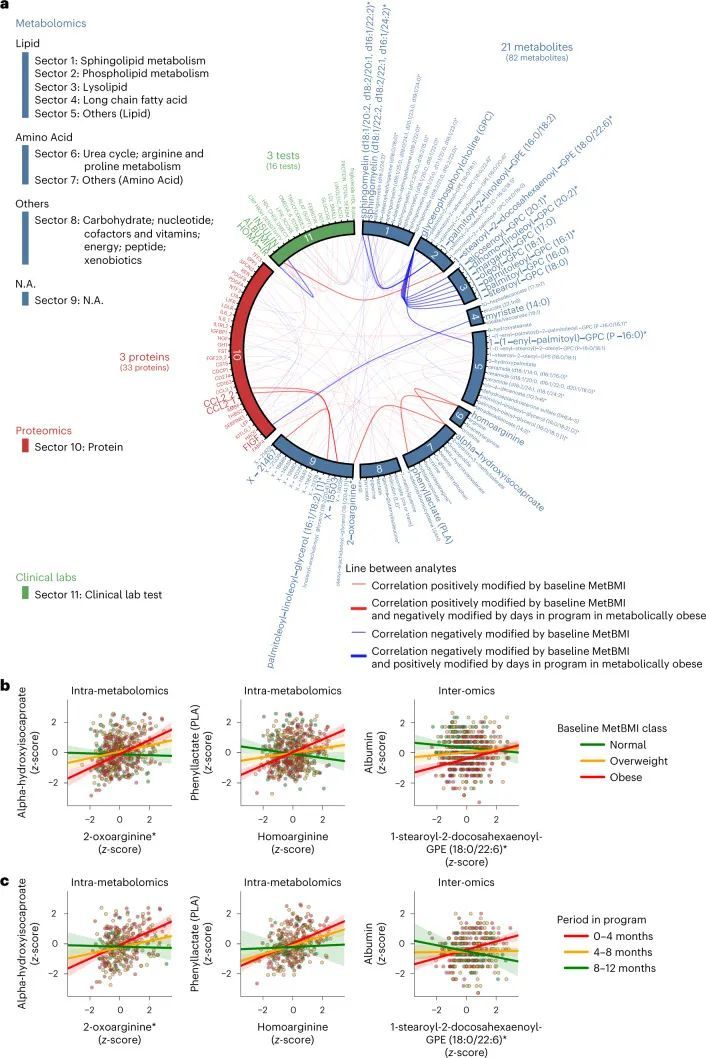
作者研究了血浆分析物相关网络的纵向变化,重点关注由MetBMI分类定义的代谢性肥胖组。基于基线代谢组学状态的重要性(图5d,e),作者首先评估了Arivale子队列(图5a;608名参与者)中每个血浆分析物-分析物相关之间与基线MetBMI之间的关系,使用广义线性模型(GLM)中的交互项来分析每个分析物-分析物对(方法)。在这种类型的模型中,统计检验评估了任意两个分析物之间的关系是否依赖于第三个变量(在本例中为基线MetBMI)。在608,856个血浆分析物的配对关系中,有100个分析物-分析物相关对,包括82种代谢物、33种蛋白质和16种临床实验室检测项目,受基线MetBMI的显著影响(FDR < 0.05;附加数据7)。随后,作者使用广义估计方程(GEE)中的交互项(即与计划中的天数的交互作用)评估了这100对在基线肥胖MetBMI组中的纵向变化(182名参与者)的每个分析物-分析物对(方法)。在100对中,有27对分析物-分析物相关对在计划的天数中显著改变(FDR < 0.05;图6a)。这27对主要来自代谢物(21个代谢物,三个蛋白质和三个临床实验室检测)。其中一个时间变化的对是同型精氨酸和苯乳酸(PLA)。同型精氨酸被发现是心血管疾病的生物标志物,并且在MetBMI和CombiBMI模型中是一个稳定的正向预测因子(图2a和扩展数据图5a)。PLA是肠道菌群产生的苯丙氨酸衍生物,具有抗菌活性和抗氧化活性。同型精氨酸和PLA之间的正相关关系在肥胖的MetBMI类别中在基线时观察到(图6b),并且在干预过程中这个类别中变得较弱(图6c),这意味着代谢异常在代谢性肥胖群体中在计划期间得到了一定程度的改善。这些发现表明代谢改善不仅限于特定血液分析物浓度的变化,还包括分析物之间的关联结构的变化。
总结
总之,这项研究强调了血液多组学分析在预测和预防医学中的实用性。它还概述了对肥胖症的前所未有的多组学特征化,并将成为评估代谢健康和确定可操作的健康管理目标的宝贵资源。
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:veading@qq.com进行投诉反馈,一经查实,立即删除!