【完整思路】2023 年中国高校大数据挑战赛 赛题 B DNA 存储中的序列聚类与比对
2023 年中国高校大数据挑战赛
赛题 B DNA 存储中的序列聚类与比对
任务
1.错误率和拷贝数分析:分析“train_reads.txt”和“train_reference.txt”数据集中的错误率(插入、删除、替换、链断裂)和序列拷贝数。
2.聚类模型开发:开发一个模型来聚类“train_reads.txt”中的序列,评估准确性(包括聚类数量和纯度)和聚类速度。
3.在测试数据上的应用:将开发的模型应用于“test_reads.txt”,这是来自不同合成环境的文件。提供聚类时间、目标序列数和拷贝数分布图。
4.比较模型开发:设计一个模型,用于比较同一聚类内的序列,以恢复原始信息。将此应用于“test_reads.txt”中的聚类序列,并输出最有可能的目标序列。
r任务1分析
文件“train_reads.txt”包含了一系列的DNA序列,每行代表一个序列。每行的格式如下:
?行首的数字表示该序列的拷贝数。
?空格后跟随的是DNA序列,由碱基(A、T、G、C)组成。
例如,第一行“75 GCGAAAGATAGTAAAGTAGCCGATTGAGTGTCCCGATTATAGGAAGTGGATCTCTTACACT”表示该序列有75个拷贝,且序列是“GCGAAAGATAGTAAAGTAGCCGATTGAGTGTCCCGATTATAGGAAGTGGATCTCTTACACT”。
文件“train_reference.txt”同样包含了一系列DNA序列,每行代表一个序列。其格式如下:
?行首的数字是序列的索引或ID。
?空格后跟随的是DNA序列,由碱基(A、T、G、C)组成。
例如,第一行“0 TAGACCCCTACACCACGTAGAAAACTCATCCTGTTCGACATGAGCTGGCCACTCCTGGAC”表示序列ID为0,序列内容是“TAGACCCCTACACCACGTAGAAAACTCATCCTGTTCGACATGAGCTGGCCACTCCTGGAC”。
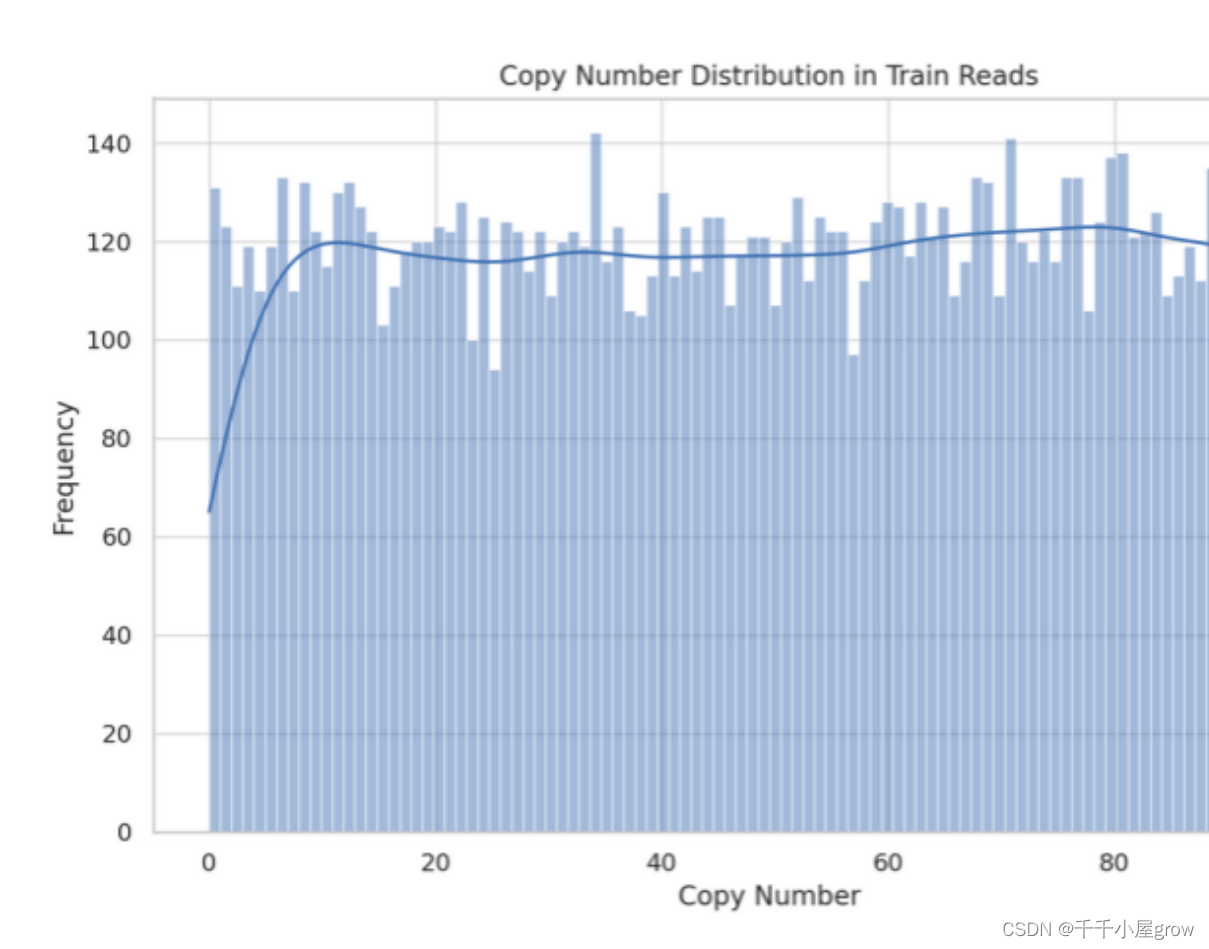
?拷贝数分布:数据集中的拷贝数分布相对均匀,覆盖了从0到近100的范围。
?频率:某些特定的拷贝数出现的频率略高于其他值,但整体上没有明显的集中趋势或偏差。
加载了“train_reads.txt”和“train_reference.txt”的数据,并将其转换为了DataFrame格式,以便于进行后续分析。数据的基本格式如下:
?
train_reads:
?
oCopy Number: 序列的拷贝数。
oSequence: DNA序列。
?
train_reference:
?
oID: 序列的索引或ID。
oSequence: DNA序列。
比较“train_reads.txt”中的序列与“train_reference.txt”中的参考序列,以识别和计算插入、删除、替换错误和链断裂的频率。
分析序列的拷贝数分布情况。
BioPython,这是一个常用于生物信息学计算的库,特别是用于DNA序列比对。因此,将使用一种不同的方法来估计序列之间的差异(包括插入、删除和替换错误)。
区分插入和删除错误,特别是当序列长度差异较大时。
没有考虑链断裂的情况。
?拷贝数(Copy Number):数据集包含100个不同的拷贝数,最常见的拷贝数是34。
?插入错误(Insertions):在12000个序列中,大多数(9610个)没有检测到插入错误。其他序列中的插入错误数量不同,共有39种不同的插入错误数。
?删除错误(Deletions):大多数序列(9535个)没有检测到删除错误,删除错误的种类相对较少,共有5种不同的删除错误数。
?替换错误(Substitutions):替换错误在序列中的分布更加广泛,共有52种不同的替换错误数,其中3727个序列没有检测到替换错误。

插入错误(Insertions)的分布
?大多数序列没有插入错误,这可以从插入错误数为0的高频率看出。
?少数序列中出现了插入错误,但这些错误的数量通常较少。
删除错误(Deletions)的分布
?同样,删除错误在大多数序列中未被检测到,表现为删除错误数为0的高频率。
?出现删除错误的序列相对较少,错误数量也较少。
替换错误(Substitutions)的分布
?替换错误的分布比插入和删除错误更广泛,表明这类错误在数据集中更为常见。
?有一定数量的序列没有替换错误,但也有很多序列中检测到了不同数量的替换错误。
任务2:聚类模型开发:开发一个模型来聚类“train_reads.txt”中的序列,评估准确性(包括聚类数量和纯度)和聚类速度。
- 数据准备
?特征提取:将DNA序列转换为适合聚类的特征。这涉及到编码序列(例如,使用k-mer频率)。
?样本选择:由于DNA序列数据量可能很大,我们可能需要选择一个代表性的样本来训练和测试聚类模型。 - 聚类模型选择
?选择算法:根据数据特性选择合适的聚类算法,如K-Means、层次聚类、DBSCAN等。
?参数调整:调整模型参数以优化聚类结果。 - 模型训练与评估
?训练模型:使用选定的算法和参数训练聚类模型。
?评估指标:
o聚类数量:确定最佳聚类数量。
o纯度:评估聚类的纯度,即每个聚类中相似序列的比例。
o聚类速度:评估模型聚类的时间效率。 - 实施与优化
?根据评估结果调整模型。
?可能需要进行多次迭代以达到最佳聚类效果。
数据降维:考虑使用主成分分析(PCA)或其他降维技术来减少特征空间的维度,从而加快聚类过程。
使用更高效的聚类算法:探索使用更适合大数据集的聚类算法,如MiniBatch K-Means或其他可伸缩的聚类方法。
优化特征提取:考虑使用不同的k-mer大小或其他特征提取方法来更有效地表达序列。
分步聚类:先在一个更小的样本集上进行聚类,然后根据这些结果调整参数和方法,再在更大的数据集上应用。
任务3:
· 在测试数据上的应用:将开发的模型应用于“test_reads.txt”,这是来自不同合成环境的文件。提供聚类时间、目标序列数和拷贝数分布图。
基于问题2,需要考虑聚类时间、目标序列数和拷贝数分布图。
任务4:
· 比较模型开发:设计一个模型,用于比较同一聚类内的序列,以恢复原始信息。将此应用于“test_reads.txt”中的聚类序列,并输出最有可能的目标序列。
目的是从可能包含错误的序列中恢复出原始信息。这个过程涉及到识别和纠正在DNA序列合成和测序过程中产生的错误。
序列预处理:由于DNA序列可能包含不同类型的错误(如插入、删除、替换),需要对序列进行适当的预处理,以便于比较。
序列比较方法:我们将开发或采用一种算法来比较同一聚类内的序列。这可能包括序列对齐、一致性评分和错误纠正。
原始信息恢复:基于比较结果,我们将尝试恢复出最可能的原始序列。这可能涉及到多数投票、概率模型或其他统计方法。
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:veading@qq.com进行投诉反馈,一经查实,立即删除!