CompAIRR
brief
CompAIRR (compairr) is a command line tool to compare two sets of adaptive immune receptor repertoires and compute their overlap.
CompAIRR可以用于比较两个免疫组库的序列,识别出哪些overlap序列(精确匹配和模糊匹配)。
It can also identify which sequences are present in which repertoires.
也可以在免疫组库中进行search,找到相同或者相似的序列。
Furthermore, CompAIRR can cluster the sequences in a repertoire set. Sequence comparisons can be exact or approximate.
对免疫组库进行聚类,实现cluster / clonotype的识别。
install
wget https://github.com/uio-bmi/compairr/archive/refs/heads/main.zip
unzip main.zip
cd compairr-main
make clean
make
make test
make install
compairr -h # successful install
compare
repertoire vs repertoire comparison
核心技术是序列的比对,包括精确比对和模糊比对,该工具支持蛋白序列和核酸序列的比对。
具体参数设置和作用如下:
-
Commands:
-h, --help display this help and exit
-v, --version display version information-m, --matrix compute overlap matrix between two sets
免疫组库之间的比较-x, --existence check existence of sequences in repertoires
免疫组库的序列检索-c, --cluster cluster sequences in one repertoire
免疫组库的聚类(形成cluster/clonotype)-z, --deduplicate deduplicate sequences in repertoires
序列去重 -
General options:
-d, --differences INTEGER number of differences accepted (0*)
序列比对时允许的编辑距离,这里的距离指的是可编辑距离(levenstand distance)
d >= 2时工具的运行速度会变慢-i, --indels allow insertions or deletions when d=1
当d >= 1 时可以设置 i参数,使LV 距离变成LCS 距离,也就是插入或者删除字符相当于距离增加
通常使用的字符距离计算方式参考上一篇记录:
https://editor.csdn.net/md/?articleId=133994192-f, --ignore-counts ignore duplicate_count information
-g, --ignore-genes ignore V and J gene information
默认是比较V/J reference gene的,该参数取消V/J reference gene必须相同的限制-n, --nucleotides compare nucleotides, not amino acids
默认是比较蛋白序列,设置为该参数则比较核酸序列(这时候 d通常会设置的比较大,运行会变慢)-s, --score STRING MH, Jaccard, product*, ratio, min, max, or mean
-t, --threads INTEGER number of threads to use (1*-256)
线程数-u, --ignore-unknown ignore sequences with unknown symbols
蛋白序列包括ACDEFGHIKLMNPQRSTVWY字符,核酸序列包括ACGTU字符,出现其他字符会报错,
除非设置该参数过滤掉包含特殊字符的序列(所以注意空白字符哦) -
Input/output options:
-a, --alternative output results in three-column format, not matrix–cdr3 use the cdr3(_aa) column instead of junction(_aa)
–distance include sequence distance in pairs file
-k, --keep-columns STRING comma-separated columns to copy to pairs file
AIRR格式的输入文件包含了很多columns,输出文件中可以设定只包括那些columns-l, --log FILENAME log to file (stderr*)
日志文件-o, --output FILENAME output results to file (stdout*)
输出文件-p, --pairs FILENAME output matching pairs to file (none*)
cat seta.tsv
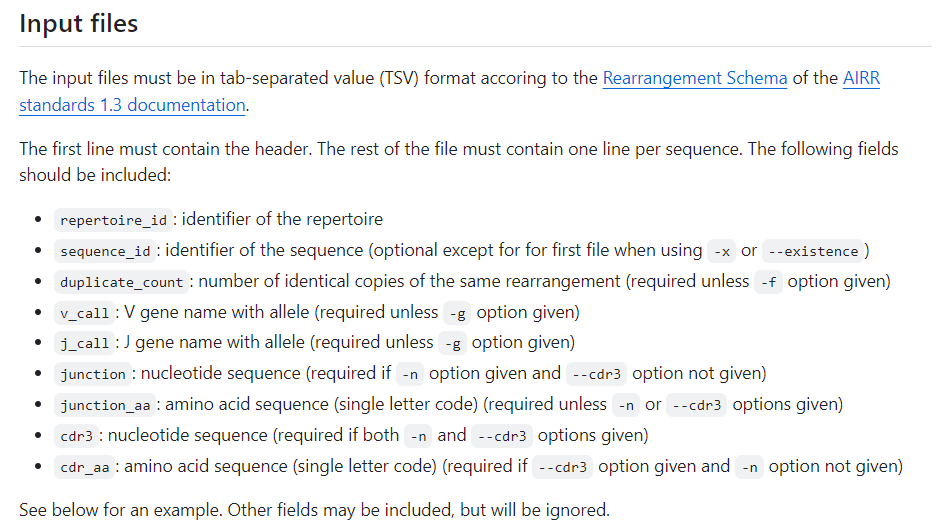
repertoire_id sequence_id duplicate_count v_call j_call junction junction_aa sequence rev_comp productive d_call sequence_alignment germline_alignment v_cigar d_cigar j_cigar
B1 T 5 TCRBV07-09 TCRBJ01-02 tgcgcgagcagcctgcgcgtgggcggctatggctataccttt CASSLRVGGYGYTF
B1 U 10 TCRBV07-09 TCRBJ01-02 tgcgcgagcagcctgcgcgtgggcggctttggctataccttt CASSLRVGGFGYTF
B2 V 7 TCRBV07-06 TCRBJ02-01 tgcgcgagcagcaccagccatcagcagtatttt CASSTSHQQYF
# To compute the overlap between two repertoire sets, use the -m or --matrix option
compairr -m seta.tsv setb.tsv -d 1 -o output.tsv -p pairs.tsv
search
# Use the option -x or --existence to analyse in which repertoires a set of sequences are present, and create a sequence presence matrix
compairr -x setc.tsv setb.tsv -d 1 -f -o output.tsv -p pairs.tsv
cluster
其实我最想用的是该命令实现聚类效果,但是该聚类效果太鸡肋了,只能死板的使用参数 d固定数字,没法根据CDR3的长度进行可编距离的变换以达到满足相似度的要求。
# To cluster the sequences in one repertoire, use the -c or --cluster option
# V/J reference 相同,CDR3 相似
compairr -c setb.tsv -d 1 -n -o output.tsv
# V/J reference 可以不相同,CDR3 相似
compairr -c setb.tsv -d 1 -n -g -o output.tsv
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:veading@qq.com进行投诉反馈,一经查实,立即删除!