2023.12.12【基因组】|bedtools与gffread序列提取比较
背景
最近在优化基因组组装流程,其中在组装完成后需要提取cds这一步。我同时试了bedtools和gffread这两个工具,得到了两个fa文件,做了一下对比,感觉还是有些差异,这里记录一下。
数据与方法
数据介绍
物种:酵母菌(真菌)
组装序列数据:Pacbio hifi数据,使用flye进行质控组装;
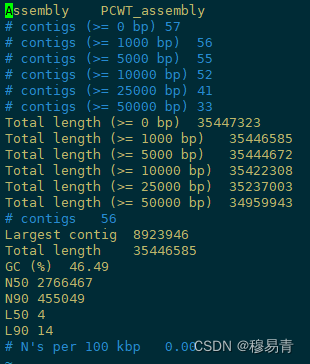
组装质量评估
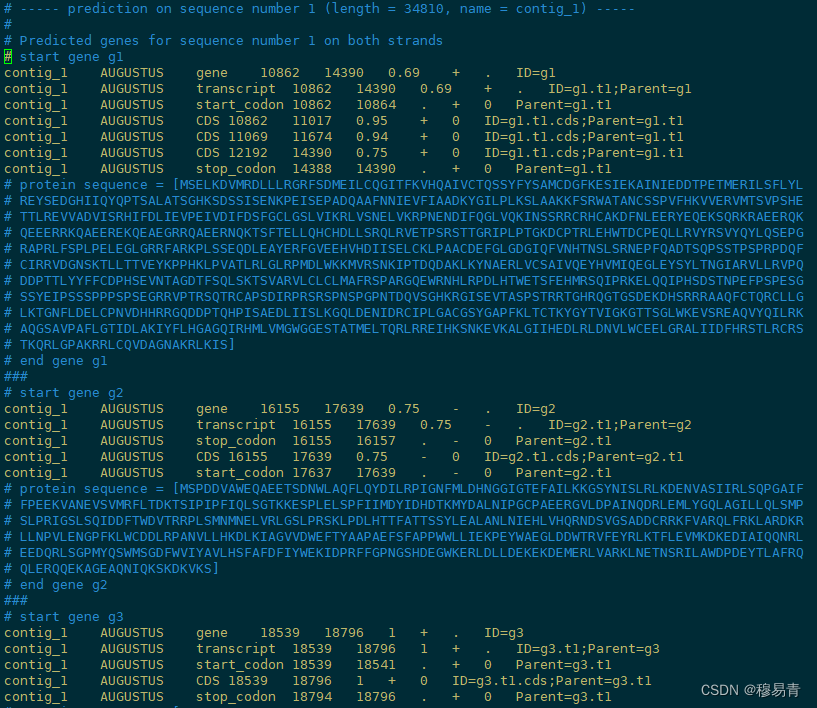
gff注释文件:使用Augustus进行注释得到gff文件,包含gene,transcript,cds,start_codon,end_codon等信息;
比较工具
bedtools
Version: v2.30.0
使用内置getfasta命令
执行命令
bedtools getfasta -fi 03.Quast/PCWT_assembly.fasta -fo 04.Annotation/PCWT/cds.fna -bed 04.Annotation/PCWT/genomic.gff
得到结果为cds.fa
gffread
案例版本未知,重新安装后为0.12.7
执行命令
gffread -w 04.Annotation/PCWT/cds2.fa -g 03.Quast/PCWT_assembly.fasta 04.Annotation/PCWT/genomic.gff
得到结果为cds2.fa
结果
提取序列结果展示
bedtools
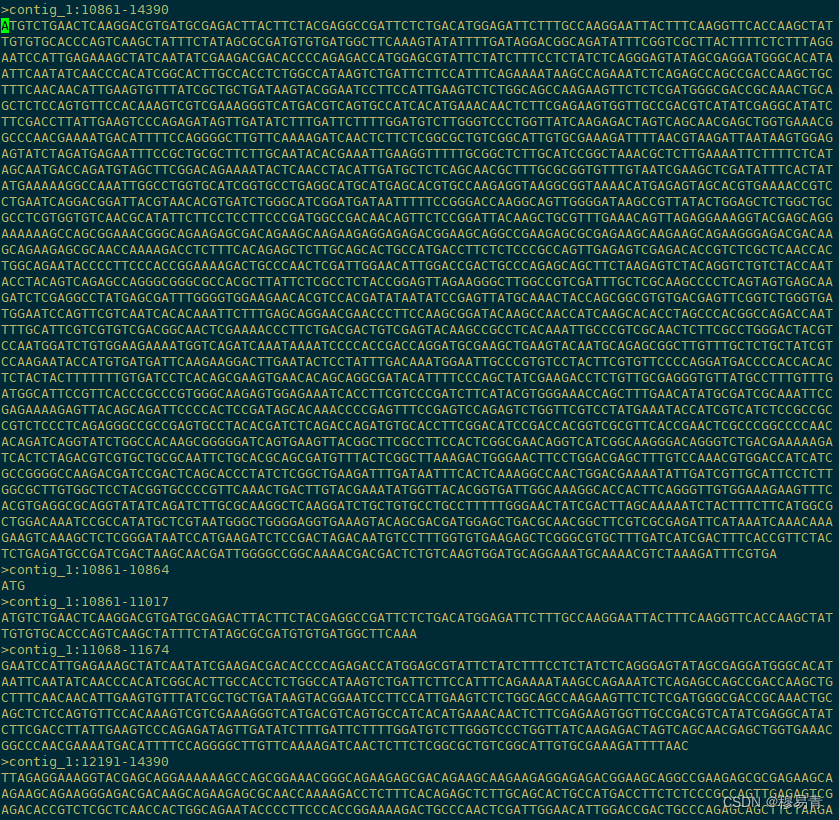
不知道是不是没有设置好参数,bedtools 读取gff文件后输出了所有序列特征(gene,transcript,cds,start_codon,end_codon),且没有比较清晰的命名情况。
gffread
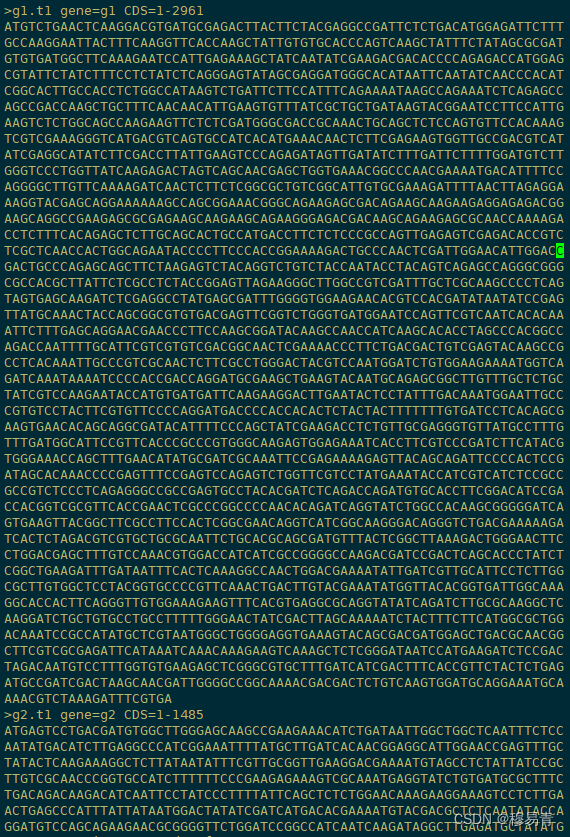
gffread输出结果要好一些,首先是标准的fasta格式(60-70bp一行),会读取cds的ID名称进行命名,只提取cds特征,不过默认参数下同一个gene下的cds是合并成一条,不会单独分开。
总结
如果对注释后gff文件的cds特征行进行筛选再进行分析,bedtools getfasta的结果也没有问题。我们平时做差异分析注释时也经常使用。将差异显著的基因信息用bed格式进行统计,然后通过bedtools getfasta从参考基因组中提取出来进行注释。不过目前了来看gffread的识别效率更高,在文章编辑时,我更新了gffread,新版本加了很多参数,功能丰富了不少,除了cds,还可以提取exon,transcript等特征序列,另外也可以支持其他注释格式(gtf,bed)。
有任何问题都可以加 vx:bbplayer2023私聊或者进群沟通,大家相互交流,共同进步。
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:veading@qq.com进行投诉反馈,一经查实,立即删除!