CD8+T细胞通过NKG2D-NKG2DL轴维持对MHC-I阴性肿瘤细胞的杀伤
今天给同学们分享一篇实验文章“CD8+ T cells maintain killing of MHC-I-negative tumor cells through the NKG2D-NKG2DL axis”,这篇文章发表在Nat Cancer期刊上,影响因子为22.7。

结果解读:
MHC-I阴性肿瘤的免疫疗法需要CD8 T细胞
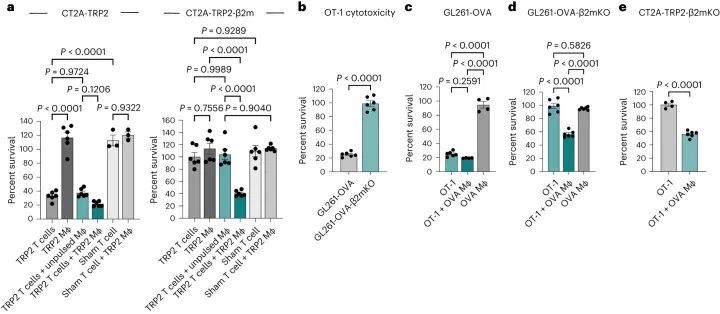
作者先前已经证明了4-1BB激动和抗程序性细胞死亡蛋白1(PD-1)检查点阻断(αPD-1/4-1BB)在小鼠CT2A胶质瘤模型中的疗效。毫不奇怪,这种疗效依赖于CD8 T细胞的存在。最近,为了研究抗原特异性反应,作者将CT2A小鼠胶质瘤细胞系改造成表达酪氨酸酶相关肽2(TRP2)的CT2A-TRP2细胞系,这是一个免疫原性较弱的模型抗原。αPD-1/4-1BB联合治疗对原位植入的CT2A-TRP2肿瘤表现出类似的疗效,导致80%的长期生存(图1a)。
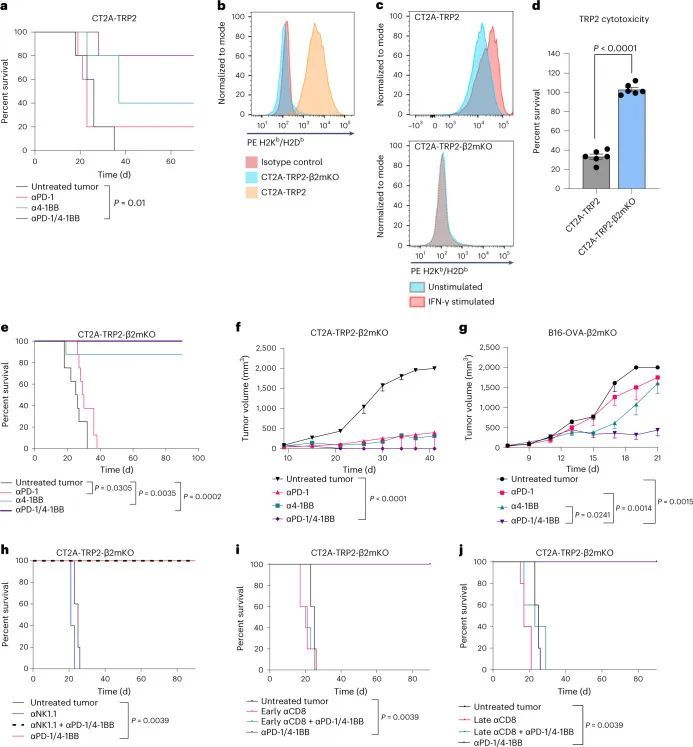
最初的目的是研究肿瘤抗原呈递对肿瘤微环境的影响,作者使用CRISPR技术在CT2A-TRP2细胞系中敲除(KO)编码β2m的基因,生成CT2A-TRP2-β2mKO细胞系(图1b)。流式细胞术检测结果显示,CT2A-TRP2-β2mKO细胞缺乏MHC-I表达(H2K和H2D)(图2)。通过干扰素(IFN)-γ刺激实验进一步确认了MHC-I的敲除。IFN-γ刺激导致亲本CT2A-TRP2细胞中H2K和H2D的上调,但CT2A-TRP2-β2mKO细胞中未见上调(图3)。此外,TRP2 TCR转导的CD8 T细胞(TRP2 T细胞)未能在体外杀死CT2A-TRP2-β2mKO肿瘤,但能有效杀死亲本CT2A-TRP2肿瘤,从而对CT2A-TRP2-β2mKO细胞系中MHC-I的敲除进行了功能验证(图4)。
令人惊讶的是,作者发现携带颅内(i.c.)CT2A-TRP2-β2mKO肿瘤的小鼠对免疫疗法仍然有反应,对αPD-1/4-1BB联合治疗表现出100%的长期生存率,对抗4-1BB单独治疗表现出90%的长期生存率(图1e)。与CT2A-TRP2肿瘤一样,未经治疗的CT2A-TRP2-β2mKO肿瘤是致命的。对CT2A-TRP2-β2mKO肿瘤的免疫治疗效果不仅限于颅内区域,αPD-1/4-1BB在皮下植入的MHC-I阴性胶质瘤中仍然有效(图1f)。此外,αPD-1/4-1BB对缺乏MHC-I的原位植入黑色素瘤(B16-F10-OVA-β2mKO,以下简称B16-OVA-β2mKO)仍然有效(图1g)。最终,MHC-I阴性肿瘤的免疫治疗成功既不局限于胶质瘤,也不局限于颅内区域。
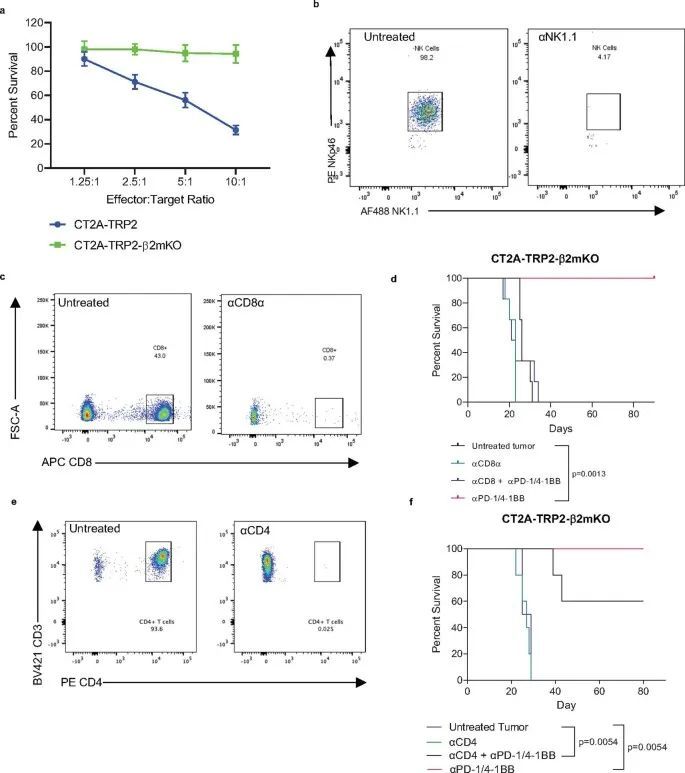
被激发了兴趣,作者试图确定免疫细胞群体对MHC-I阴性肿瘤免疫疗法的疗效负责。由于已知NK细胞能够消除缺失或减少MHC的细胞,作者首先在植入CT2A-TRP2-β2mKO肿瘤之前通过消耗NK细胞进行了调查(扩展数据图1b)。即使没有NK细胞,αPD-1/4-1BB仍然能够引发100%的长期生存(图1h)。因此,作者进一步探讨了免疫治疗对CT2A-TRP2-β2mKO肿瘤的疗效是否仍然依赖于CD8 T细胞,即使肿瘤没有MHC-I表达。消耗CD8 T细胞(扩展数据图1c)完全取消了αPD-1/4-1BB对携带CT2A-TRP2-β2mKO肿瘤的小鼠的生存益处。这一点在肿瘤植入之前(扩展数据图1d)或肿瘤植入后早期(第3天)或晚期(第7天)时间点(图1i,j)消耗CD8 T细胞都是成立的。值得注意的是,CD4 T细胞的消耗对生存率有一定但不显著的影响(扩展数据图1e,f)。
特异性抗原杀伤在MHC-I阴性肿瘤中持续存在
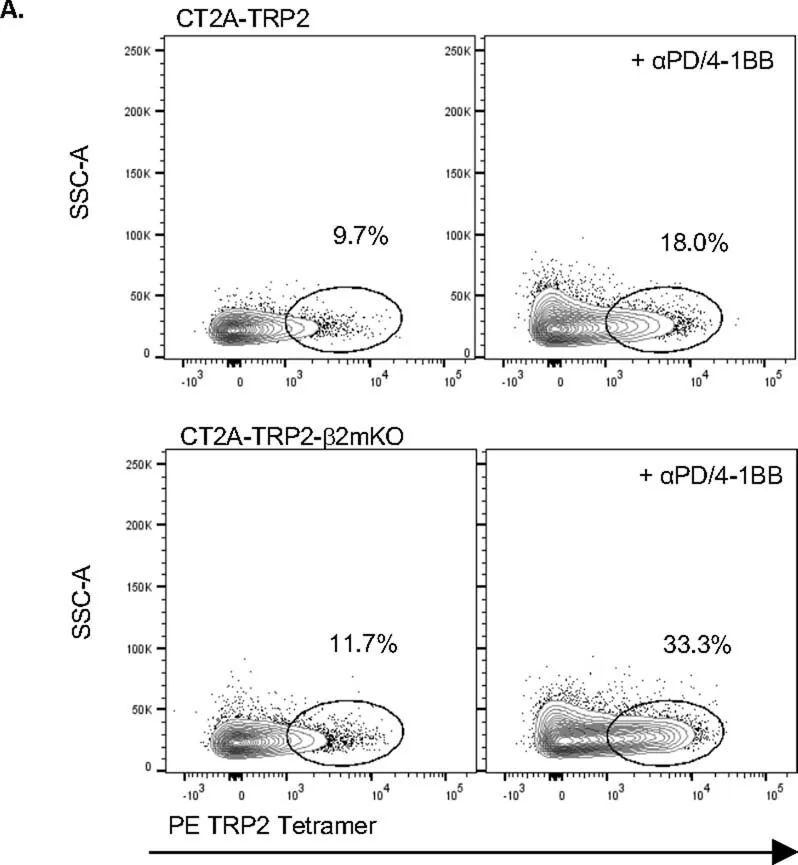
继续依赖CD8 T细胞引发了一个问题,即是否还以特异性抗原的方式对MHC-I阴性肿瘤进行细胞毒杀。有趣的是,作者注意到αPD-1/4-1BB治疗的MHC-I阴性CT2A-TRP2-β2mKO肿瘤的小鼠体内,TRP2特异性CD8 T细胞的数量比同样接受治疗的MHC-I阳性CT2A-TRP2肿瘤的小鼠体内更多(图2a和扩展数据图2a)。为了测试抗原特异性的作用,作者将CT2A-TRP2-β2mKO或CT2A-β2mKO肿瘤(缺乏TRP2和MHC-I表达)植入缺乏CD8 T细胞(CD8KO)的小鼠的脑内。随后,小鼠接受TRP2特异性CD8 T细胞的移植(TRP2 ALT),产生一个完全特异于TRP2的T细胞组分(图2b)。TRP2 ALT与αPD-1/4-1BB联合治疗足以消除100%的CT2A-TRP2-β2mKO肿瘤(图2c),但对缺乏TRP2表达的CT2A-β2mKO肿瘤则完全无效(图2d)。此外,作者将CT2A-TRP2-β2mKO肿瘤植入转基因OT-1小鼠的脑内。这些小鼠中的大多数T细胞识别卵清蛋白(OVA)肽段(OVA 257–264 ,SIINFEKL)的残基257-264,但对TRP2没有反应。在这些小鼠中,αPD-1/4-1BB的组合对CT2A-TRP2-β2mKO肿瘤完全无效(图2e),进一步表明即使在没有肿瘤MHC-I抗原呈递的情况下,肿瘤特异性T细胞仍然是必需的。
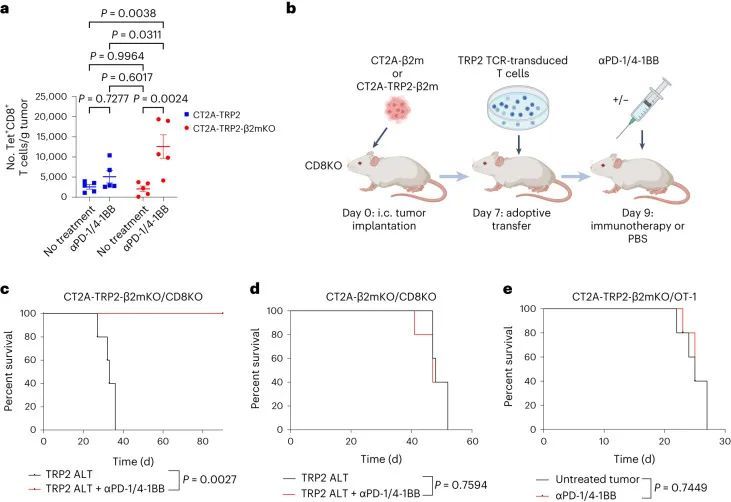
上面作者证明了TRP2 T细胞无法在体外杀死CT2A-TRP2-β2mKO肿瘤细胞(图1d),这证实了单独的CD8 T细胞无法杀死MHC-I阴性肿瘤,即使存在相应的抗原。因此作者假设在体内观察到的细胞毒性必须由其他细胞群体协助。在体内,浸润的髓系细胞构成了脑肿瘤微环境中的大部分免疫细胞。因此,作者检查了是否添加骨髓源性巨噬细胞(BMDMs)可以让TRP2 T细胞在体外杀死CT2A-TRP2-β2mKO肿瘤。CT2A-TRP2或CT2A-TRP2-β2mKO(图3a)肿瘤细胞与TRP2 T细胞一起培养,同时加入TRP2肽负载的BMDMs(TRP2巨噬细胞)或未负载的BMDMs(未负载巨噬细胞)。虽然TRP2 T细胞单独(但不是TRP2巨噬细胞)能够有效杀死MHC-I阳性的CT2A-TRP2肿瘤,但是TRP2 T细胞单独或TRP2巨噬细胞单独都无法杀死MHC-I阴性的CT2A-TRP2-β2mKO肿瘤。然而,令人惊讶的是,TRP2巨噬细胞和TRP2 T细胞的组合确实足以在体外杀死CT2A-TRP2-β2mKO肿瘤。有趣的是,未经脉冲处理的巨噬细胞与TRP2 T细胞的组合也未能导致对肿瘤细胞的显著杀伤,这表明巨噬细胞与CD8 T细胞之间的抗原-同源相互作用对于对缺乏MHC-I的肿瘤细胞产生细胞毒性是必要的。虚假的TCR转导的CD8 T细胞无法杀死任何一种肿瘤细胞系,即使在TRP2负载的巨噬细胞存在的情况下也是如此。重要的是,作者使用OVA特异性OT-1 T细胞和OVA脉冲的巨噬细胞在另一种表达不同靶抗原(OVA)的小鼠胶质瘤细胞系(GL261)中重复了这些发现(图3b-d)。
CD8 T细胞通过直接接触杀死MHC-I阴性肿瘤
作者接下来研究了杀死缺乏MHC-I的肿瘤细胞的机制要求。首先确定是否涉及可溶性因子,作者使用了0.4微米的Transwell板,它允许细胞因子和其他可溶性介质自由通过,但不允许细胞通过。将MHC-I阴性的CT2A-TRP2-β2mKO肿瘤细胞放置在Transwell板的底部,TRP2 T细胞和TRP2巨噬细胞的不同组合被添加到膜的同侧或对侧。将T细胞和/或巨噬细胞与肿瘤细胞物理分离消除了肿瘤杀伤活性,表明需要直接细胞间接触而不是可溶性因子(图4a)。

然后作者调查了巨噬细胞或T细胞是否更直接地参与了接触依赖性肿瘤细胞毒杀机制。为了进行这些实验,作者使用了5.0微米的Transwell板,这样较小的T细胞可以通过到含有肿瘤细胞的孔底部,而较大的巨噬细胞则无法通过(验证见扩展数据图3a)。CT2A-TRP2-β2mKO肿瘤细胞被放置在Transwell插入物的对面,与T细胞和巨噬细胞隔开。这种设置允许T细胞首先与巨噬细胞直接接触,然后通过膜与肿瘤细胞直接接触。在这种情况下,肿瘤杀伤的程度与三种细胞类型都接触时没有显著差异(图4a)。这表明,杀伤MHC-I阴性肿瘤需要肿瘤细胞与TCR刺激的T细胞直接接触,而巨噬细胞只需要提供前驱抗原特异性T细胞刺激的角色。

NKG2D介导T细胞对MHC-I阴性肿瘤的识别
为了阐明CD8 T细胞识别MHC-I阴性肿瘤的细胞-细胞接触依赖机制的性质,作者首先采用无偏方法,研究了抗原特异性CD8 T细胞在暴露于MHC-I阴性肿瘤和相应抗原负载的巨噬细胞时的炎症基因表达谱的差异。将与OVA脉冲的巨噬细胞共培养的经过激活的OT-1 T细胞的基因集与仅与OVA阴性CT2A-TRP2-β2mKO肿瘤细胞共培养的经过激活的OT-1 T细胞进行比较。使用OVA阴性的β2mKO肿瘤细胞系以确保肿瘤-TCR相互作用的缺失。这些实验中的OT-1细胞的TCR激活是通过与OVA脉冲的巨噬细胞同时培养来完成的。接触CT2A-TRP2-β2mKO细胞的TCR激活的OT-1细胞表达了标志NK和T细胞功能的基因,而仅仅是TCR激活的OT-1细胞没有这种增加(图5a)。在T细胞功能基因集中,不同表达的基因包括编码激活标志的基因,但不包括直接接触介导的细胞毒性的受体(扩展数据图4a). 值得注意的是,在凋亡基因集中,Tnfsf10的表达略有增加,该基因编码TNF相关凋亡诱导配体(TRAIL)(log 2 (折叠变化)= 0.562,调整后的P值= 0.0262)(扩展数据图 图4b)。因此,考虑到其在接触依赖性T细胞细胞毒性中的已知作用,作者调查了TRAIL是否参与杀死体外MHC-I阴性肿瘤。然而,阻断TRAIL并没有显著减少T细胞介导的体外细胞毒性,无论是针对MHC-I阳性还是MHC-I阴性肿瘤(扩展数据图 图4c)。

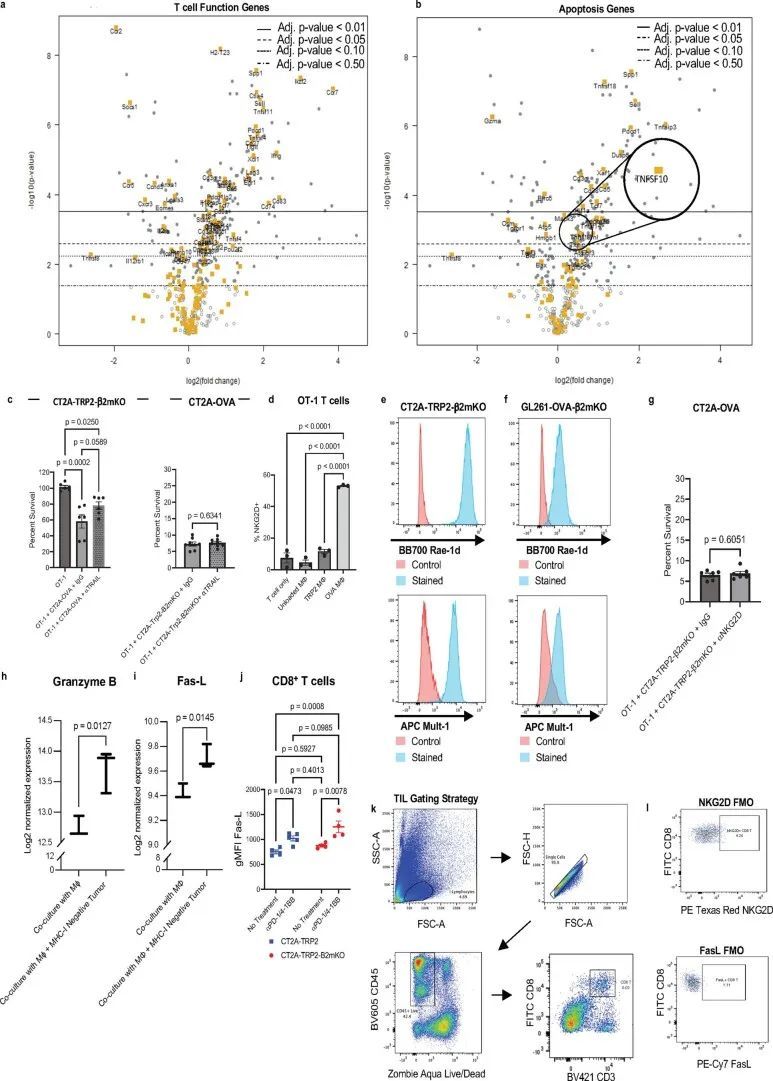
在NK细胞功能基因集中,Klrk1的表达在暴露于CT2A-TRP2-β2mKO细胞的OT-1细胞中特别突出(图5b,log(折叠变化)= 1.1,调整的P值= 3.25×10)。NKG2D是一个激活受体,传统上与NK细胞的细胞毒功能相关,尽管它也在活化的CD8 T细胞上表达,其作用主要被描述为共刺激。在小鼠中,NKG2D的配体是β2m非依赖的非经典MHC-I分子RAE-1、H60和MULT-1。与在所有有核细胞上表达的经典MHC-I不同,非经典MHC-I的表达通常是由应激诱导的,并且在肿瘤细胞上上调。已经显示肿瘤细胞上非经典MHC-I的表达在接受化疗或电离辐射后进一步上调。
同样地,在TCR激活后,NKG2D受体与靶细胞上的配体结合,激活NKG2D细胞中的细胞毒性效应,包括释放细胞毒性颗粒和表达Fas配体(FasL)。特别是在CD8 T细胞中,NKG2D细胞效应似乎依赖于同时的TCR激活,以将效应活性限制在适当的周围靶标上。在小鼠CD8 T细胞中,NKG2D表达在TCR激活后上调(扩展数据图4d)。由于作者模型中CD8 T细胞介导的MHC-I阴性肿瘤杀伤也依赖于TCR激活,但不需要T细胞与肿瘤细胞之间的抗原配对相互作用,作者假设NKG2D可能是肿瘤杀伤机制的重要因素。为了初步评估这一点,作者首先检查了模型中T细胞上的NKG2D表达。因此,作者检查了未经处理的小鼠中浸润CT2A-TRP2或CT2A-TRP2-β2mKO胶质瘤的CD8 T细胞上NKG2D表达的差异,以及经αPD-1/4-1BB治疗的小鼠中的差异。NKG2D水平在治疗的MHC-I阴性CT2A-TRP2-β2mKO肿瘤浸润的CD8 T细胞上明显高于治疗的MHC-I阳性肿瘤或未经处理的肿瘤(图5c)。检测到T细胞上NKG2D水平的增加,作者反过来检查了两种MHC-I阴性肿瘤系的相关NKG2DLs的存在。发现CT2A-TRP2-β2mKO系高度表达非经典MHC-I分子RAE-1和MULT-1,这些发现在额外的胶质瘤系GL261-OVA-β2mKO中得到了重复(扩展数据图4e,f)。
为了研究NKG2D对CD8 T细胞对MHC-I阴性肿瘤的功能贡献,作者重复了作者之前的体外肿瘤细胞毒杀实验,这次在有或没有NKG2D阻断抗体的情况下进行。OT-1 T细胞再次与OVA阴性、MHC-I阴性的CT2A-TRP2-β2mKO肿瘤细胞培养,尽管在这些实验中,作者使用CT2A-OVA细胞作为OVA抗原呈递和OT-1 TCR激活的来源。与作者之前的结果一样,只有在有OVA呈递细胞的情况下,OT-1 T细胞才能成功杀死CT2A-TRP2-β2mKO肿瘤(图5d)。然而,在存在抗NKG2D阻断抗体的情况下,对CT2A-TRP2-β2mKO细胞的杀伤完全被抑制(图5d)。使用CT2A-OVA肿瘤作为OVA TCR刺激的来源还使作者能够评估抗NKG2D治疗对MHC-I阳性肿瘤细胞的伴随杀伤的影响。在任何条件下,CT2A-OVA细胞都能够被轻易杀死(扩展数据图4g)。
NKG2D信号传导的下游细胞毒性效应物可以包括颗粒酶、穿孔素和FasL29,31,这就提出了一个问题,即在作者的系统中,其中哪些可能介导MHC-I阴性肿瘤细胞的杀伤。作者的表达分析数据显示,颗粒酶B和FasL表达增加(扩展数据图4h,i)在暴露于MHC-i阴性肿瘤的TCR激活的T细胞中,表明脱颗粒可能在抑瘤机制中发挥作用。事实上,与单独的巨噬细胞相比,在MHC-I阴性肿瘤细胞和携带同源抗原的巨噬细胞存在的情况下,OT-1 T细胞的脱颗粒增加(图5e)。鉴于先前研究的结果32-34,作者还专门测试了FasL-Fas相互作用的作用。肿瘤浸润性CD8+T细胞上FasL的表达在免疫治疗的反应中增加,但在从MHC-I阳性肿瘤和MHC-I阴性肿瘤中分离的CD8+T淋巴细胞中没有显著差异(扩展数据图4j–l)。此外,在体外,抗原阴性Fas受体KO(FasKO)黑色素瘤仍然易被TCR激活的CD8+T细胞杀死(扩展数据图图5a),表明FasL-Fas相互作用对肿瘤抑制机制并不重要。

总结
在这里,作者描述了MHC-I和抗原非依赖性CD8+T细胞杀伤肿瘤细胞的机制,该机制在体内和人类肿瘤细胞中一致可见。作者证明,CD8+T细胞依赖性免疫疗法确实可以对肿瘤保持有效,即使在完全缺乏MHC-I的情况下也是如此。需要进一步研究NKG2D和NKG2DL在介导肿瘤免疫治疗易感性中的作用,以及这些发现的其他潜在治疗意义。对这篇文章的思路感兴趣的老师,欢迎咨询!
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:veading@qq.com进行投诉反馈,一经查实,立即删除!