第一性原理——模拟计算
2023-12-22 17:46:17
?第一性原理计算的基本思想是将多个原子构成的体系看成是由多个电子和原子核组成的系统,并根据量子力学的基本原理对问题进行最大限度的“非经验性”处理。它只需要5个基本常数(m0,e,h,c,kB)就可以计算出体系的能量和电子结构等物理性质。它可以确定已知材料的结构和基础性质,并实现原子级别的精准控制,是现阶段解决实验理论问题和预测新材料结构性能的有力工具。并且,第一性原理计算不需要开展真实的实验,极大地节省了实验成本,现已被广泛应用于化学、物理、催化、环境、生命科学和材料等领域。
适合的研究方向包括但不限于:金属材料、非金属材料、纳米材料、半导体材料、电催化光催化、热催化、电池、固体、界面、合金、吸附等
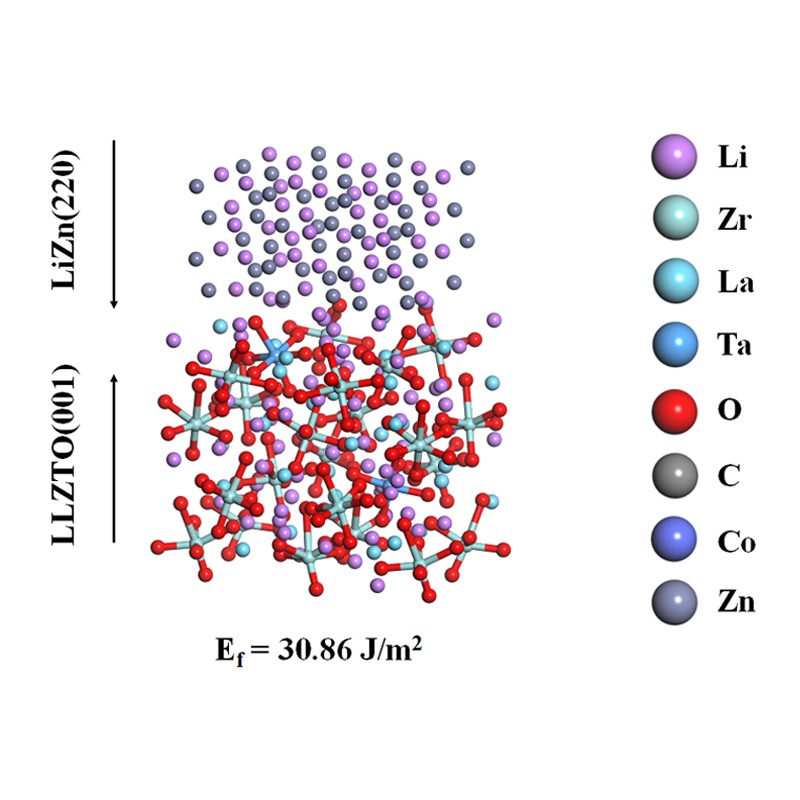
可以计算的体系包括但不限于:晶体、非晶、二维材料、表面、界面、固体等
常用软件:常用软件:VASP,Materials Studio (MS),CP2K,Quantum Espresso (QE),Gaussian,Wannier90等
可以计算的内容包括但不限于:?
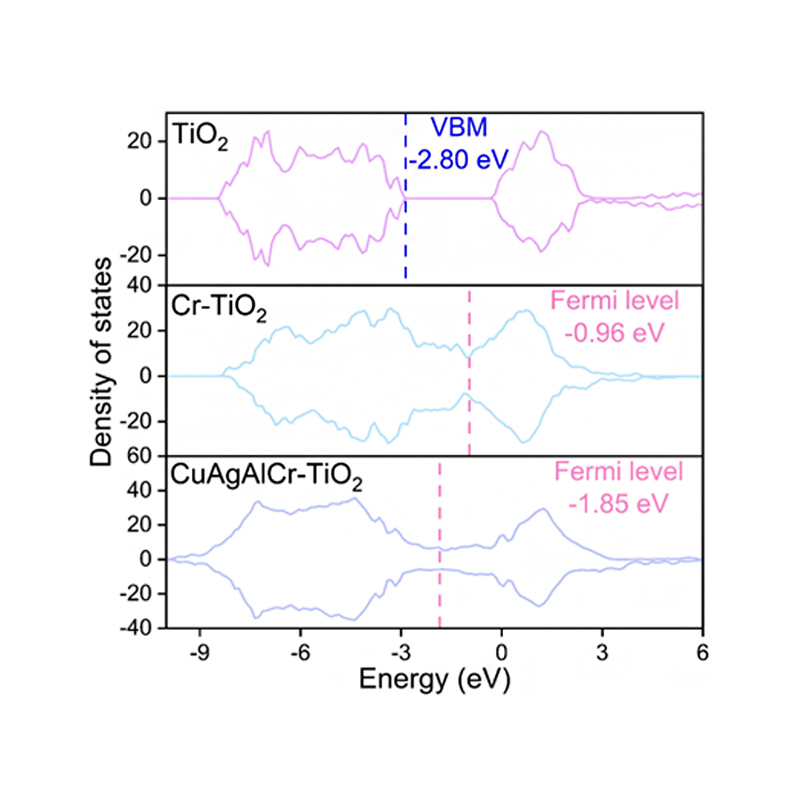
1、电子结构计算,如电荷密度、电荷差分密度、态密度、能带、费米能级、功函数、ELF等?
2、几何结构计算,如键长、键角、二面角、晶格常数、原子位置等
3、材料性质计算,如介电常数、弹性模量、磁导率、热导率、界面热阻等
4、能量相关计算,如吉布斯自由能、吸附能、掺杂能、形成能等
5、反应相关计算,如反应路径、反应机理研究、过渡态搜索、能垒计算等
6、其他计算,如声子谱、异质结、锂-硫电池、碱金属离子电池、高熵合金计算等
 ?
?
如果有需求可以联系我喔

文章来源:https://blog.csdn.net/weixin_70563937/article/details/135155439
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:veading@qq.com进行投诉反馈,一经查实,立即删除!
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:veading@qq.com进行投诉反馈,一经查实,立即删除!